Geoscience at the nanometre scale:
review of analytical transmission electron microscopy applications
B. M. PetrunicDepartment of Geology, University of New Brunswick, PO Box 4400, Fredericton, New Brunswick, E3B 5A3, Canada; *Corresponding Author
petrunic@gmail.com
T. A. Al
Department of Geology, University of New Brunswick, PO Box 4400, Fredericton, New Brunswick, E3B 5A3, Canada
L. C. Cavé
Microscopy and Microanalysis Facility, University of New Brunswick, PO Box 4400, Fredericton, New Brunswick, E3B 5A3, Canada
V. J. Banks
Department of Geology, University of New Brunswick, PO Box 4400, Fredericton, New Brunswick, E3B 5A3, Canada
D. B. Loomer
Department of Geology, University of New Brunswick, PO Box 4400, Fredericton, New Brunswick, E3B 5A3, Canada
C. S. Belfry
Microscopy and Microanalysis Facility, University of New Brunswick, PO Box 4400, Fredericton, New Brunswick, E3B 5A3, Canada
S. R. Cogswell
Microscopy and Microanalysis Facility, University of New Brunswick, PO Box 4400, Fredericton, New Brunswick, E3B 5A3, Canada
L. Weaver
Microscopy and Microanalysis Facility, University of New Brunswick, PO Box 4400, Fredericton, New Brunswick, E3B 5A3, Canada
Date received: 15 December 2005 ¶ Date accepted: 13 September 2006
ABSTRACT
This paper describes applications of analytical Transmission Electron Microscopy (TEM) in the geosciences. The topics include: 1) sulphide-mineral oxidation; 2) trace-metal attenuation by secondary Mn oxides; 3) silicate weathering; 4) transition-metal valence in minerals; and 5) secondary Hg minerals in stream sediments. The main advantage of the analytical TEM is the ability to obtain images, chemical information, and electron diffraction patterns at the nanometre scale. With such high spatial resolution, it is possible to observe physical and chemical features in samples that cannot be resolved with most other techniques. This information can lead to significant improvement in our understanding of the system under investigation. Sample preparation techniques that are used in each study are also described in this paper. The preparation of samples for TEM analysis can be challenging because of the heterogeneity commonly encountered in geological materials, the fragility of some geological samples (e.g., low-temperature minerals), and the need to maintain spatial relationships present in the samples. The sample preparation techniques presented are specific to the needs of the study and the appropriateness of these methods is demonstrated by the high quality analytical TEM data that are obtained.RÉSUMÉ
Cet exposé décrit des applications de la microscopie électronique à transmission analytique dans les sciences de la terre. Les aspects étudiés comprennent : 1) l'oxydation des minéraux sulfurés; 2) l'atténuation des métaux-traces par des oxydes de Mn secondaires; 3) la silicatisation météorique; 4) la valence des métaux de transition dans les minéraux; et 5) les minéraux de Hg secondaires dans les sédiments fluviatiles. Le principal avantage qu'offre la MET analytique est la possibilité d'obtenir des images, des données chimiques et des figures de diffraction des électrons à l'échelle nanométrique. Une résolution spatiale aussi élevée permet l'observation dans les échantillons de propriétés physiques et chimiques impossibles à éclaircir au moyen de la majorité des autres techniques. De tels renseignements peuvent mener à une amélioration marquée de notre compréhension du système à l'étude. Cet exposé décrit en plus les techniques de préparation des échantillons utilisées lors de chaque étude. La préparation des échantillons à une analyse MET peut s'avérer compliquée en raison de l'hétérogénéité que présentent communément les matières géologiques, de la fragilité de certains échantillons géologiques (p. ex. minéraux à basse température) et de la nécessité de maintenir les liens spatiaux présents dans les échantillons. Les techniques de préparation des échantillons présentées sont propres aux besoins de l'étude; les données de haute qualité obtenues des analyses MET témoignent de la pertinence de ces méthodes. [Traduit par la rédaction]INTRODUCTION
1 In recent years, research at the nanometre (10-9 m) scale has grown rapidly because of the recognition that many physical and chemical properties of materials are significantly different at this scale compared to the macroscopic or microscopic scale. Discoveries of new and unexpected properties at the nanoscale are leading to a better understanding of physical and chemical processes, and in some cases these discoveries are resulting in significant economic benefits. The ability to view and analyze materials at this scale has led to advances in computer and medical technology. Nanoscience is at the frontier of knowledge partly because of the difficulty associated with conducting detailed studies of materials at this scale. Applications of nanoscale analyses in the geosciences have previously been summarized by Hochella (2002a,b). In general, the argument for conducting geological studies at the nanoscale is based on the idea that larger scale measurements may not provide a true representation of the system of interest.
2 Analytical Transmission Electron Microscopy (TEM) is one of the most common and versatile techniques that allows for scientific investigations at the nanoscale. Although TEM has long been used in biology, geology, and materials engineering, the potential use of TEM in the geosciences has recently expanded because of the development of sophisticated new analytical systems that operate in the TEM, and new technologies that are well-suited for the preparation of geological samples. Analytical TEM is a versatile tool because samples can be well-characterized at the nanoscale using a combination of imaging, analytical, and electron diffraction techniques. We have employed analytical TEM in a variety of geological applications, and the overall objective of this paper is to summarize some of the results with the aim of demonstrating the applicability of analytical TEM to a range of geological topics.
3 One of the primary advantages of analytical TEM, including Scanning TEM mode (STEM) in which the beam rasters over the sample, is the ability to image a sample at much higher magnification compared to commonly used techniques in geological studies such as the Scanning Electron Microscope (SEM) and Electron Microprobe (EMP). For instance, it is common to obtain images using the (S)TEM at magnifications that exceed 100, 000 times. In addition, a variety of different imaging techniques are available making the TEM a versatile imaging tool. For instance, Centred Dark Field (CDF) imaging can be used to illuminate areas in the sample that have a consistent crystallographic orientation, Annular Dark Field (ADF) imaging can be used to illuminate regions that are strongly diffracting, and High-Angle Annular Dark Field imaging (HAADF) generates contrast on the basis of spatial variations in atomic number (Z). Only a brief summary of some (S)TEM imaging techniques is provided here, but readers are referred to Williams and Carter (1996) for more specific information.
4 In addition to imaging, chemical information can be obtained in the (S)TEM using Energy-Dispersive X-ray Spectroscopy (EDS) and Electron Energy-Loss Spectroscopy (EELS). Energy-Dispersive X-ray Spectroscopy is the measurement of characteristic X-rays that are emitted as the electron beam interacts with the sample, thereby providing information about the chemical composition of the material. This technique is undoubtedly familiar to SEM users, but unlike in the SEM, the collection of quantitative EDS analyses in the (S)TEM is far less routine. Quantitative (S)TEM-EDS analyses are problematic because homogeneous standards for use at the nanoscale are not readily available, and it is difficult to obtain measurements on standards and samples under similar instrument operating conditions. The main advantage of collecting EDS analyses in the (S)TEM compared to the SEM, is the ability to obtain analyses at a much greater spatial resolution. It is possible to obtain STEM– EDS analyses with a spatial resolution of less than 5 nm in the plane of section, where the spatial resolution in this mode is governed by the type of electron source, the beam size, and the beam spreading in the sample (interaction volume) (Williams and Carter 1996).
5 Electron Energy-Loss Spectroscopy is another analytical technique that is commonly used in the (S)TEM. This technique involves the measurement of characteristic energy loss in the transmitted electron beam that results from electron interactions with the sample. In general, EELS spectra can provide more information than EDS regarding the chemical nature of the material. Similar to EDS, EELS data provide information about the chemical composition of the material, but the spectra can also be used to determine the chemical bonding environment and valence for chemical components of the material (Egerton 1996). The spectra obtained by STEM– EELS are similar to those obtained using Near Edge X-ray Absorption Fine Structure (NEXAFS) spectroscopy; however, the spatial resolution of STEM-EELS is better than the spatial resolution of X-ray absorption spectroscopy, which is approximately 50 nm using a Scanning Transmission X-ray Microscope (STXM) (Benzerara et al. 2005; Braun et al. 2005; Hitchcock et al. 2005). In contrast, the energy resolution of STXM– NEXAFS surpasses that of STEM– EELS (Rightor et al. 1997; Braun et al. 2005).
6 Finally, site-specific electron diffraction patterns can be collected at the nanoscale using Selected-Area Electron Diffraction (SAED), microdiffraction, and/or Convergent Beam Electron Diffraction (CBED) techniques. Patterns obtained with these techniques can provide information about the mineralogy and structural properties of the sample, which can be extremely useful when chemical analyses are unable to uniquely identify the material (e.g., in the case of polymorphic minerals).
7 The ability to combine various imaging, analytical, and diffraction techniques make the (S)TEM an extremely useful tool for analyzing geological materials that can be heterogenous at the nanoscale; however, sample preparation is a major hurdle that has stood in the way of TEM studies of geologic materials. The primary concern when preparing samples for (S)TEM analysis is ensuring that the samples are electron transparent, which means they must be extremely thin (Williams and Carter 1996). The exact thickness that is required varies depending on the material, but samples are generally less than a few hundred nanometres thick for the collection of EDS and diffraction data, and less than 100 nm thick for the collection of EELS data. An ideal sample would also be non-magnetic, electrically conductive, of uniform thickness, and stable under the electron beam. Although most samples are not ideal, many different sample preparation techniques are available, and instrument parameters may be adjusted to accommodate the analysis of most materials. Some of the challenges that are commonly encountered in the preparation of geological specimens include: 1) the need to preserve spatial relationships (e.g., between adjacent grains at a reaction boundary); 2) the preparation of samples that contain both hard and soft components (e.g., soft coating on a hard mineral); and 3) the requirement to avoid chemical, physical, and mineralogical changes during sample preparation (e.g., heat-induced mineralogical changes; dissolution of mineral phases).
8 In this paper, the following geological topics are used to illustrate the applicability of the analytical (S)TEM in the geo-sciences: 1) sulphide-mineral oxidation; 2) mineral formation and trace-metal attenuation during in situ chemical oxidation; 3) low-temperature silicate weathering; 4) measurement of transition-metal valence in minerals; and 5) low-temperature Hg mineral formation. The specific (S)TEM analysis techniques that were used to understand these processes will be presented in the sections that follow. In addition, specific sample preparation techniques for TEM analysis for each topic will be fully described within each section. This paper is a review of work that has been previously described and references to these publications can be found within the individual sections.
INSTRUMENTATION
9 Analytical (S)TEM was carried out using a JEOL 2011 instrument with a LaB6 cathode at 200 kV. Energy Dispersive X-ray Spectroscopy analyses were collected with an EDAX Genesis 4000 EDS System and EELS analyses were collected with a post-column Gatan Image Filter (GIF2000). Several instruments were used to prepare samples including a Micrion 2500 Focused Ion Beam (FIB) instrument, a Leica Ultracut UCT ultramicrotome equipped with a diamond knife, and a Gatan Model 691 Precision Ion Polishing System (PIPS).
GEOSCIENCE APPLICATIONS
Sulphide-Mineral Oxidation
Background
10 Sulphide-mineral oxidation is commonly cited as the cause of poor water quality at mine sites worldwide because it results in acid-mine drainage, which is the release of acidity, sulphate, and dissolved metals (e.g., Alpers and Blowes 1994; Jambor and Blowes 1994; Jambor et al. 2003). Therefore, understanding sulphide-mineral oxidation is an important step toward predicting the geochemical evolution of drainage water from tailings and implementing mitigation measures that will ensure environmentally acceptable mine closure. One of our research interests is related to the oxidation of arsenopyrite (FeAsS) in the tailings from the Mount Pleasant Tungsten Mine, which is located approximately 60 km south of Fredericton, NB. The geochemistry, hydrogeology, and mineralogy of the Mount Pleasant Mine tailings has been previously presented by Petrunic and Al (2005), and from this study it is evident that elevated As concentrations occur in the near-surface pore water (maximum of 7.1 mg/L). The abiotic and biotic oxidation of arsenopyrite under a variety of conditions (e.g., air and aqueous solutions) has been studied previously (e.g., Buckley and Walker 1988; Richardson and Vaughan 1989; Nesbitt et al. 1995; Fernandez et al. 1996a, b; Nesbitt and Muir 1998; Schaufuss et al. 2000; Jones et al. 2003; Mikhlin and Tomashevich 2005). Many of these studies have used spectroscopic techniques to detect changes in oxidation state for As, Fe, and S as arsenopyrite oxidizes and secondary minerals form. From previous studies, it is evident that arsenopyrite oxidation leads to the formation of various As- and/or Fe-bearing secondary minerals. The aim of this study is to build on previous research by employing analytical (S)TEM to study secondary minerals that have formed as a result of arsenopyrite oxidation and interactions with the local pore-water chemistry. The results from this work are summarized here but are explained in detail by Petrunic et al. (2006).
TEM sample preparation
11 The loose tailings were collected using aluminum casing (7.5 cm diameter), which was later cut in half lengthwise. The tailings were then dried and impregnated with low-viscosity epoxy. Double-polished thin sections were prepared from the epoxy-impregnated blocks. Oxidized arsenopyrite grains were identified in the thin sections using SEM in the backscattered electron mode, and the FIB was used to prepare two sections for (S)TEM analysis from specified locations at the margins of two arsenopyrite grains (Fig. 1A). The FIB sample preparation technique was chosen for this study because it is possible to prepare site-specific sections from minerals of contrasting hardness while maintaining spatial relationships. The FIB method described by Patterson et al. (2002) was used to prepare samples in this study. It was possible to obtain a sample from each grain at a specified location, with the section extending from the epoxy-filled pore space, through the soft secondary-mineral coating, and into the relatively hard arsenopyrite grain (Fig. 1B). In this way, the spatial relationship between the pore space, the coating, and the arsenopyrite was maintained, allowing for a meaningful interpretation of the results. Although FIB was initially developed for materials science, it is a useful tool for the preparation of high quality specimens for (S)TEM analysis from a wide variety of geological samples (e.g., Heaney et al. 2001; Dobrzhinetskaya et al. 2003; Lee et al. 2003; Seydoux-Guillaume et al. 2003; Wirth 2004; Benzerara et al. 2005).
Fig. 1 A) Backscattered SEM image of an oxidized arsenopyrite grain. The rectangle represents the location where the sample for TEM analysis was prepared using a FIB instrument. B) A low magnification TEM image of the sample prepared from the location shown in A. The interface between the arsenopyrite grain and the coating is indicated by the solid line. The dotted square indicates the approximate location of the image shown in (C). C) High-Angle Annular Dark Field image (HAADF) image showing the relative location of mineral phases that were identified in the coating.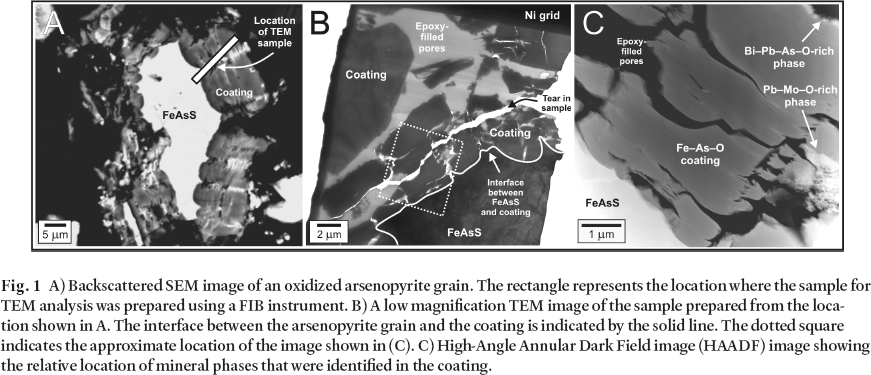
Summary of results
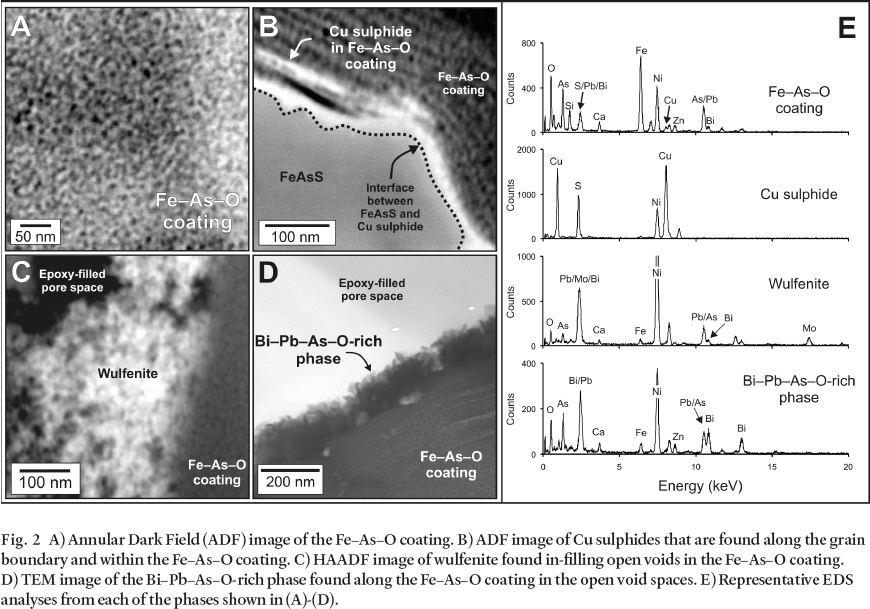
12 The secondary coating extends several microns from the arsenopyrite grain boundary (Fig. 1B). A higher magnification HAADF image acquired from the rectangular area outlined in Fig. 1B, demonstrates the heterogeneity of the coating (Fig. 1C). In this image it is possible to note the presence of several distinct phases which were characterized by EDS analyses. These phases include an Fe– As– O coating, a Cu– S-rich phase, a Pb– Mo– O-rich phase, and a Bi– Pb– As– O-rich phase. Examples of each of these phases are presented at still higher magnification in Figs. 2A-D, and representative STEM– EDS analyses are shown in Fig. 2E. Unique mineral identification requires chemical as well as structural information, therefore, electron diffraction patterns were collected from each of the phases in either SAED and/or microdiffraction mode and these patterns were compared against simulated electron diffraction patterns of known minerals. These analyses indicate that the Fe– As– O coating is amorphous, the Cu– S-rich phase is crystalline and the patterns are consistent with chalcocite (Cu2S) and/ or djurleite (Cu31S16), the Pb– Mo– O-rich phase is crystalline and the patterns are consistent with wulfenite (PbMoO4), and the Bi– Pb– As– O-rich phase is crystalline but the diffraction patterns could not be indexed conclusively.
13 Based on imaging, EDS, and electron diffraction data collected from the two samples, it was concluded that arsenopyrite oxidation leads to the precipitation of the amorphous Fe– As– O coating. This is consistent with previous studies, which have identified a range of As- and (or) Fe-rich coatings as arsenopyrite oxidation products (Buckley and Walker 1988; Richardson and Vaughan 1989; Nesbitt et al. 1995; Nesbitt and Muir 1998; Schaufuss et al. 2000). The main difference between this and previous studies arises from the ability to collect images, chemical analyses, and diffraction data from the same sample at the nanoscale while preserving spatial relationships. This allows for a better understanding of the influence of the pore-water composition on secondary-mineral formation. With the exception of the Fe– As– O coating, the formation of the secondary minerals that are observed requires the input of additional elements (Cu, Pb, Mo, Bi) to the local geochemical system. The addition of these elements is accomplished by advection or diffusion of aqueous solutes in the pore space adjacent to the arsenopyrite grain. For instance, in addition to finding discrete Cu sulphides disseminated in the bulk coating, Cu sulphides were observed along the arsenopyrite grain boundary. This suggests that a reaction between aqueous Cu(II) and arsenopyrite occurs in the tailings, resulting in the precipitation of a Cu sulphide along the boundary according to the following simplified reaction (scorodite represents the Fe– As– O coating):

Display large image of Figure 2
14 Continued oxidation of arsenopyrite and retreat of the grain boundary leads to the isolation of Cu sulphides in the Fe– As– O coating in the form of layers aligned parallel with the arsenopyrite grain boundary (Fig. 2B) and aggregates (not shown). The precipitation of wulfenite and the Bi– Pb– As– O-rich phase occurs in the open voids within the Fe– As– O coating in response to changes in saturation state for these phases in the local pore-water.
15 More details regarding the processes mentioned above can be found in Petrunic et al. (2006), but the overall conclusion is that there is a complex assemblage of secondary minerals in this system and their compositional and mineralogical characteristics can only be adequately assessed at the nanometre scale.
Mineral formation/trace-metal attenuation during in situ chemical oxidation
Background
16 Chlorinated solvent contamination is common in ground-water systems, resulting from subsurface accumulations of immiscible solvents which are part of a larger group of Dense Non-Aqueous Phase Liquids known as DNAPLs. Trichloroethylene (TCE; C2Cl3H) and tetrachloroethylene (also known as perchloroethylene or PCE; C2Cl4) are two of the most widely used solvents. A common approach to aquifer restoration is the in situ destruction of chlorinated ethane sources using chemical oxidation, referred to as In Situ Chemical Oxidation (ISCO). The permanganate ion (MnO4 -) is a strong oxidant that is frequently employed in the form of either the potassium or sodium salt (KMnO4 and NaMnO4). The injection of MnO4 - into aquifers, and subsequent reactions with TCE and PCE, results in profound changes in the redox state and may lead to a large decrease in pH as well:


17 The reduction of Mn(VII), the overall increase in the aquifer redox state, and the addition of acidity to the aquifer trigger a variety of mineral-water interactions in the aquifer. The two most important reactions are the precipitation of insoluble Mn oxides with Mn valence ranging between II and IV (herein referred to generally as MnOx), and pH buffering from dissolution of carbonate minerals (Nelson et al. 2001; Huang et al. 2002; Crimi and Siegrist 2004; Li and Schwartz 2004). Aquifer systems commonly contain small amounts of heavy metals that are naturally bound in a variety of stable mineral forms. The radical changes in redox and pH conditions that accompany ISCO could lead to the mobilization of these metals. The acid-generating and pH-buffering reactions represent important controls on trace-metal solubility and adsorption behaviour (Pretorius and Linder 2001; Kanungo et al. 2004; Tonkin et al. 2004).
18 One of the objectives of this study was to use analytical (S)TEM to investigate metal sequestering by MnOx that form during ISCO in a controlled laboratory column experiment. The input solution to the sand-packed columns contained trace metals (Cu, Pb, Zn, Ni, Mo) in addition to MnO4 and TCE. A flow gradient was induced along the columns and the reaction was monitored under calcite (CaCO3) buffered and unbuffered conditions. The results that pertain to the carbonate-buffered system are summarized here, but readers are referred to Al et al. (2006a) for a more detailed description of the research.
TEM sample preparation
19 The MnOx that form as a result of ISCO are expected to coat the silica and calcite sand grains present in the columns. This process presents a problem because it can be very challenging to prepare samples for TEM analysis that contain both hard and soft components such as MnOx-coated silica grains. Although the thin section/FIB method described earlier could have been suitable for this study, the columns could not be dismantled to allow sampling for thin-section preparation because the columns were required in a second experiment. Therefore, a technique that would minimize disturbance to the system was necessary. Although hard minerals can be successfully sectioned by ultramicrotomy using a diamond knife, damage to the knife can easily occur, making the technique costly. As an alternative to these methods, we mixed polypropylene threads with the silica and calcite sand during construction of the columns so that they could be removed later through side ports installed along the length of the columns. As the KMnO4— TCE reaction proceeded and MnOx formed, the oxides coated the threads. As an artificial substrate, the threads were easily sectioned with a diamond-knife mounted on an ultramicrotome. Prior to ultramicrotomy, the MnOx coated threads were embedded in a resin block immediately after extracting them from the column to avoid mineralogical changes due to drying and desiccation. NANOPLAST® FB101 was used to embed the threads because it can accommodate small amounts of water during curing.
Summary of results
20 The MnOx coatings were characterized by analytical (S)TEM. In the plane of section, the coatings occur as fibrous masses that extend outward, generally normal to the surface of the thread (Fig. 3A). The fibres are surrounded by finer-grained, interstitial MnOx that has no discernable crystal habit. Although the fibrous habit within the coatings suggests that there is some ordered structure, diffraction patterns obtained by SAED suggest that the material is amorphous.
21 Analyses of the coatings by EDS display detectable Si, Mo, Fe, Ni, and Cu (Fig. 3B). Note that the large Cu peaks in spectra 1-3 of Fig. 3B are artifacts of the Cu-mesh grids used to support the samples in the TEM. Similarly, the large Ni peaks in spectra 4 and 5 are artifacts from Ni grids. Despite these artifacts, Ni is detected in the samples that were supported on Cu grids (spectra 1-3) and Cu is detected in samples supported on Ni grids (spectra 4 and 5).
22 The general conclusion from the (S)TEM analyses is that secondary MnOx formed as a result of ISCO is capable of sequestering a variety of metals either through adsorption and/or co-precipitation mechanisms. The MnOx is extremely fine grained and intergrown with oxides and hydroxides of Fe, Si, and Al, and it is therefore not possible with techniques other than (S)TEM to obtain the discrete analyses necessary to support this conclusion.
Low-temperature silicate weathering
Background
23 Groundwater conditions are typically reducing in fractured crystalline rocks at depths of 500 to 1000 m, potentially providing a stable geochemical environment for waste isolation as proposed in the deep geologic repository concept of nuclear waste management. However, over tens of thousands of years, the glaciation events that are expected would cause changes in the hydrogeological and geochemical environment possibly leading to the ingress of dissolved O2 to greater depths than at present. Paleohydrogeological evidence for the depth of penetration of oxygenated fluids within fractured crystalline shield environments in the past may provide insight into future changes due to variations in climate and groundwater flow conditions.
24 Conceptually, the movement of oxygenated groundwater through a fracture zone leaves geochemical evidence in the form of a zone of alteration around the fracture. The strongest alteration occurs in minerals at the fracture surface where advectively transported oxygen oxidizes the reduced species, especially Fe2+, at the mineral surfaces. As a result, secondary Fe3+-bearing minerals are often precipitated on the edges of the fracture. A diffusive flux of oxygen away from the fracture into the matrix causes a wider zone of oxidation extending for some distance into the wall rock adjacent to the fracture. Oxygen diffuses along the grain boundaries between minerals, reacting first with the mineral surface and then slowly diffusing into the grain. Minerals in this region may show zonation from reacted rims to unreacted cores, producing a trend of decreasing Fe3+/ ∑Fe with distance into the mineral. Reactions with oxygen and water may also destabilize the crystal lattice of primary minerals, such as biotite (K(Mg,Fe)3(AlSi3)O10(OH)2), leading to the formation of secondary clay minerals in the alteration zone. The aim of this study was to demonstrate the use of analytical TEM in the identification of oxidative alteration products in biotite samples obtained from a rock core adjacent to a fracture in the Lac du Bonnet granite batholith. More details regarding this study can be found in Cavé and Al (2005).
Fig. 3 A) Bright-field TEM (BF TEM) image of Mn-oxide minerals that form a coating on the polypropylene thread (upper left corner). The polypropylene thread was taken from the carbonate-containing sand column at a distance of 5 cm from the inlet. B) Representative STEM– EDS analyses from the Mn-oxide coatings. The spectra were collected with a 5 nm probe while rastering the beam over a predefined area of the specimen for 200 to 500 seconds. Spectra 1-3 were collected from the areas defined in (A), the locations for Spectra 4 and 5 are not shown. The large Cu peaks in spectra 1-3, and the large Ni peaks in spectra 4-5 are artifacts from the use of Cu and Ni specimen-support grids.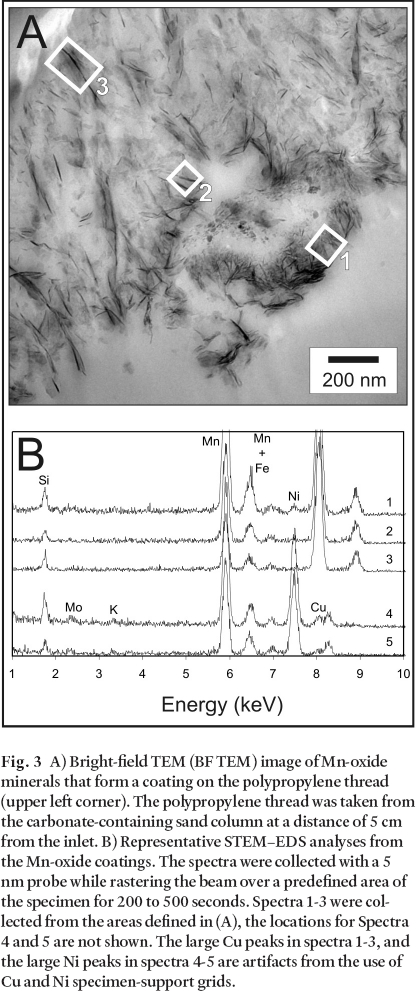
TEM sample preparation
25 The Precision Ion Polishing System (PIPS) was used to prepare samples for TEM analysis. This technique is a routine method for the preparation of geological samples. The general steps to obtaining such samples include the identification of regions of interest in a standard 30 µm double-polished petrographic thin section mounted on a glass slide with CrystalbondTM thermo polymer adhesive, the attachment of a standard 3 mm diameter TEM grid (aperture or slot grid) over the region of interest, and thinning the sample to electron transparency in the PIPS.
Summary of results
26 Biotite grains located at various distances from the fracture were prepared for (S)TEM analysis. Figure 4A presents an ADF image of a biotite grain at an intermediate magnification that was found 5 mm from the fracture. It is evident from the variable contrast in the image that the material is not homogeneous. In an attempt to explain the inhomogeneity, additional characterization was done using EDS mapping, thereby providing compositional images (Figs. 4B-H) from the same area as the ADF image (Fig. 4A). Consistent with the ADF imaging, the EDS elemental mapping reveals that on the submicron scale, the composition of biotite adjacent to the oxidized fracture is not uniform, as was suggested by observations made at the micron scale using EMP (McMurry and Ejeckam 2002) and SEM– EDS (Cavé and Al 2005) analyses.
27 The original biotite (K(Mg,Fe,Ti)3(AlSi3)O10(OH)2), contains Ti and K which are lost along with some of the Fe as the mineral alters to vermiculite (Mg0.33(Mg,Al,Fe3+)3(AlSi3)O10(OH)2). The final stage of alteration is manifest by the presence of kaolinite (Al2Si2O5(OH)4) and iron oxyhydroxide. The Fe map (Fig. 4B) highlights areas of high Fe content between 50 and 100 nm thick, which are Si and Al poor, representing Fe oxyhydroxide layers identified by SAED patterns as goethite (FeO(OH)). The Al and Si maps (Figs. 4E, 4G) contain a small wedge-shaped region in the lower left with elevated Al, Si, and O only, representing kaolinite.
28 The low temperature weathering of igneous biotite generally follows the sequence (Dong et al. 1998; Jeong and Kim 2003):

29 The weathering products identified in Fig. 4 provide evidence for oxidative alteration of the biotite. The aluminosilicate alteration products of biotite represent robust indicators of past redox changes because, unlike Fe oxyhydroxides, they are not susceptible to reductive dissolution if the system later reverted to reducing conditions.
30 This study was a test of concept, and it was known that these samples had undergone oxidative alteration. Although the alteration is not evident at the scale of SEM and EMP investigations, the STEM imaging and EDS elemental mapping techniques demonstrate that these alteration products are detectable at the nanoscale.
Measurement of transition-metal valence in minerals
Background
31 Iron is the fourth most abundant element in Earth's crust. Iron-bearing minerals are found in a large variety of geologic environments, ranging from high pressure and temperature deep within the crust and mantle, to low temperature and pressure at Earth's surface. In Earth's crust, Fe occurs mainly in two valence states, ferrous (Fe2+) and ferric (Fe3+), and it cycles between these states in response to prevailing reduction-oxidation (redox) conditions. Consequently, measurements of Fe valence ratios (Fe3+/∑Fe) in minerals can provide a useful measure of past redox states in geologic systems. For example, measurements of Fe3+/∑Fe in minerals and silicate glasses can be used to investigate oxygen fugacity in the mantle (McCanta et al. 2004; Williams et al. 2004; Bezos and Humler 2005), and estimate temperature and pressure conditions for igneous and metamorphic rocks (Schumacher 1991; Andersen et al. 1991; Holdaway et al. 1997; Wu et al. 2004). In low temperature environments, measurements of the Fe3+/∑Fe ratio may provide a tool for estimating the depth of infiltration of oxygenated groundwater into bedrock, which has applications in fields such as climate change studies and the safe siting of geological repositories for nuclear waste disposal.
32 Traditional methods of quantifying Fe3+/∑Fe have involved wet chemical analysis, Mössbauer spectroscopy, or calculations from microprobe data using mineral stoichiometry and charge balance considerations. One of the challenges of these bulk methods has been the problem of spatial resolution, especially in fine-grained materials with spatial heterogeneity. Advances in X-ray microbeam and synchrotron methods have allowed Fe3+/∑Fe measurements to be made on individual mineral grains using techniques such as X-ray Photoelectron Spectroscopy (XPS) (Raeburn et al. 1997), X-ray absorption spectroscopy (Cressey et al. 1993; Dyar et al. 2002) and Synchrotron Mössbauer Spectroscopy (SMS) (Jackson et al. 2005). However, the best spatial resolution to date for measuring Fe3+/∑Fe ratios has been obtained from EELS in the TEM (Garvie and Buseck 1998; van Aken et al. 1998; van Aken and Liebscher 2002; Cavé et al. 2006).
Fig. 4 (Facing Page) STEM– EDS elemental maps used to identify biotite and its alteration products in a portion of the sample located 5 mm from the fracture. The ADF image (A) displays visible contrast among the mineral phases which are identified with the aid of the elemental maps.
Display large image of Figure 4
33 The main objective of this research was to develop a method for one- and two-dimensional mapping of Fe valence in oxide minerals using coupled STEM– EELS. Modified from the procedures developed by van Aken et al. (1998), the Fe3+/∑Fe ratios are measured by a calibrated relationship between Fe valence and Fe L3/L2 white line intensity ratios in EELS spectra. The method is demonstrated with spectrum images of the Fe3+/∑Fe changes occurring across mineral interfaces between ilmenite (Fe2+TiO3) and magnetite (Fe2+Fe3+2O4) in a sample with microscale exsolution lamellae. A summary of the results is presented here but readers are referred to Cavé et al. (2006) for more details.
TEM sample preparation
34 Iron valence measurements were calibrated using Fe-bearing mineral standards of known Fe valence: fayalite (Fe2+2SiO4), magnetite, and hematite (Fe3+2O3). Samples of ilmenite, goethite (Fe3+O(OH)) and a different sample of hematite were analyzed as unknowns to verify that the method produced reliable results for samples of known Fe valence. Grain mounts of the calibration standards were examined by SEM and analyzed by SEM– EDS to confirm their purity.
35 Mineral standards were prepared for STEM– EELS by grinding to a fine powder in an agate mortar and pestle. Only a small amount of fayalite was available, so the crystals were crushed to powder between glass slides. Crushed material was suspended in 100% ethanol and sonicated. The suspension was withdrawn during sonication using a micropipette and dropped on a holey carbon coated molybdenum TEM grid. The standards were lightly carbon coated to prevent specimen drift during TEM analysis. This method provides areas on the edges of crushed grains which are thin enough for EELS analysis.
36 An intergrown magnetite/ilmenite sample from the University of New Brunswick Mineral Collection (Sample UNB45-1078) was used for Fe valence mapping. This magnetite was a different sample from that used as a calibration standard. A double-polished, 30 µm petrographic thin section was prepared on a glass slide, areas of finely intergrown magnetite and ilmenite were identified under reflected light, and the PIPS method of preparing samples for TEM analysis was used. Following thinning, samples of magnetite/ilmenite were carbon coated.
Summary of results
37 The sample of finely intergrown magnetite and ilmenite (Fig. 5) was ideal for developing Fe valence mapping and testing the spatial sensitivity of the technique because the ex-solution lamellae provide sharp boundaries between zones of markedly different Fe valence states. Ilmenite lamellae in the sample range in width from 1 to >100 µm. Data for the average composition of ilmenite suggest that there is substitution of Mn2+ and Mg2+ for Fe2+ and that about 10% of the Ti4+ sites are occupied by Fe3+. Numerous irregularly-shaped inclusions of spinel (MgAl2O4) were evident from optical microscopy and SEM, particularly within the ilmenite. Trace quantities of Al, Ti, and Cr may substitute within the magnetite structure, but these elements could also be present in the form of spinel and ilmenite inclusions, which are not resolved at the scale of the SEM– EDS analysis.
38 Point determinations of Fe3+/∑Fe by STEM– EELS were made on magnetite and ilmenite before acquiring spectrum images. Magnetite Fe3+/∑Fe values cover a small range, with a mean of 0.67 ± 0.03 (mean ± 1ó), which is in agreement with the theoretical value for magnetite. In contrast, STEM– EELS determinations of Fe3+/∑Fe ratios for ilmenite are more scattered, ranging between 0 and 0.20. The STEM– EELS valence measurements on ilmenite are consistent with calculations of Fe3+/∑Fe based on SEM– EDS analyses and mineral stoichiometry, which suggest that the average Fe3+/∑Fe ratios are 0.18 ± 0.02.
Fig. 5 A) BF TEM image of the area where the line scan data were collected. B) Measurements of Fe3+/∑Fe for an 80 point line scan across the ilmenite/magnetite interface shown in (A). Error bars indicate the variations in valence ratios based on one standard deviation of the measured L3/L2 ratios for magnetite. The dark solid line indicates a five-point moving average through the Fe3+/∑Fe data.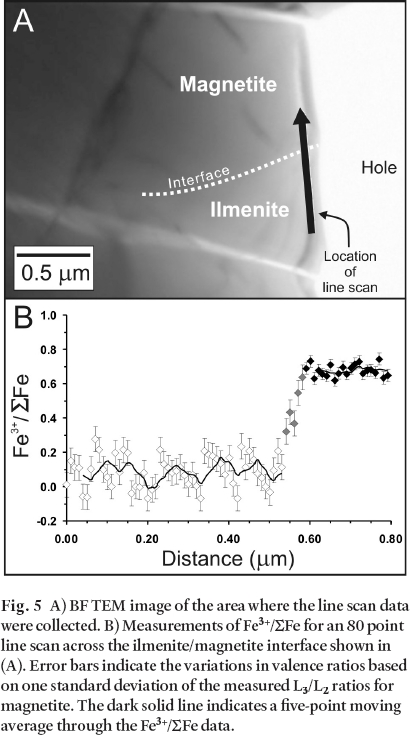
Display large image of Figure 5
39 An example of a Fe3+/∑Fe line scan, obtained using the STEM– EELS imaging method developed in this study, is shown in Fig. 5. Line scan data are presented as a scatter plot of Fe3+/∑Fe versus distance along the path shown in Fig. 5A. This method has also been used to acquire 2D analytical images of the Fe valence across the magnetite/ilmenite interface (Cavé et al. 2006). The data demonstrate that the Fe valence in magnetite is relatively constant, suggesting that in this magnetite sample there is no substitution of Fe2+ in the Fe3+ sites or vice versa. Variations in the Fe3+/∑Fe for magnetite may arise from substitutions in the magnesioferrite-jacobsite-magnet-ite mineral series (Mg2+Fe3+2O4 – (Mn,Fe,Mg)2+(Fe,Mn)3+2O4 – Fe2+Fe3+2O4), but this specimen does not contain sufficient Mg or Mn to have a significant effect on the Fe3+/∑Fe. There is a measurable variation in Fe3+/∑Fe within the ilmenite grains, with fluctuations between 0 and 0.20 over submicron distances. Substitution of Fe3+ is allowed within the ilmenite lattice. At high temperatures, there is a continuous solid-solution series between ilmenite and the Fe3+ end-member, hematite, which is achieved by coupled substitution of Fe3+ for Ti4+ and Fe3+ for Fe2+ (Andersen et al. 1991).
40 Intermediate Fe3+/∑Fe values were found for 5 points along the interface region between the two minerals in the line scan (Fig. 5). Since no attempt was made to ensure that the mineral interface intersects the sample plane at 90°, it is likely that the intermediate values reflect a mixed ratio from overlap of the minerals along an inclined interfacial boundary plane. When the 10 nm probe approaches very close to the grain boundary, the electron beam passing through the sample may interact with material on both sides of the interface to produce a weighted average signal for the two minerals.
41 The collection of STEM– EELS data as described in this study provides an extremely useful method of obtaining quantitative Fe valence measurements at the nanoscale, which can be applied to many geological issues. A similar approach has been taken to determine the Mn oxidation state in minerals and this is discussed in detail by Loomer et al. (2007).
Low-temperature Hg mineral formation
Background
42 The Murray Brook volcanogenic massive-sulphide deposit in northern New Brunswick consists mostly of pyrite and Cu — Zn— Pb sulphides, and it is overlain by a Cu-rich supergene-enrichment zone and a surficial gossan (Boyle and Smith 1994; Boyle 1995). The gossan is composed primarily of goethite, quartz (SiO2), beudantite (PbFe3(AsO4)(SO4)(OH)6), jarosite ((K,Na)Fe3(SO4)2(OH)6), plumbojarosite (PbFe6(SO4)4(OH)12), argentojarosite (AgFe3(SO4)2(OH)6), bindheimite (Pb2Sb2O6 6 (O,OH)) and scorodite with accessory cinnabar (HgS), native Ag, Au, and cassiterite (SnO2) (Boyle 1995). The gossan was mined by open pit methods between 1989 and 1992. The ore was crushed, treated with cement and hydrated lime, and sprayed with Ca— CN solution for Au/Ag extraction. The CN leaching process also extracted Hg from the ore, and a total of 1000 kg of Hg were recovered during the Au-production process. The tailings, which contained residual Hg and CN, were stored on a prepared pad of compacted glacial till (Lemon and Pheeney 1996).
43 Residual CN in the tailings causes mobilization of Hg to the groundwater beneath the tailings in the form of Hg-CN complexes (Boyle and Smith 1994; Shaw et al. 2006), which in turn discharges into the headwaters of Gossan Creek. Gossan Creek, a first-order stream in the Upsalquitch River catchment contains highly elevated concentrations of Hg (Maprani et al. 2005). Previous work by Leybourne et al. (2000) indicated that residual CN has also been responsible for the mobilization of Au from the tailings to the creek via the groundwater flow system.
44 Despite ongoing efforts to understand the geochemistry and ecotoxicology of Hg, there has been relatively little research effort to identify the discrete mineral hosts for Hg that accumulate in sediment in response to contamination events. However, this information is necessary for the evaluation of long-term environmental impacts related to bioavailability and leachability. Numerous authors present the results of analyses that indicate high concentrations of Hg in stream sediment proximal to Hg mines and other sources of Hg contamination (Gosar et al. 1997; Lechler et al. 1997; Frazier et al. 2000; Gray et al. 2002; Gray et al. 2004). Gray et al. (2004) report that some of the elevated Hg in the stream sediments at Azogado Creek in Spain can be explained by the presence of detrital cinnabar and calcine material that has been transported to the stream from waste piles. Similarly, Lechler et al. (1997) report the presence of Au– Hg amalgam and elemental Hg in sediments, and suggest that these were released directly during mining activities near Carson River, Nevada. Results of chemical extractions provide indirect evidence that Hg may partition from water to the organic and inorganic fractions of sediments (Lechler et al. 1997; Drexel et al. 2002). Kim et al. (2004a,b) make use of X-ray absorption spectroscopy to demonstrate the adsorption of Hg onto Fe- and Al-hydroxides, and Barnett et al. (1997), using electron microscopy techniques, identified metacinnabar in Hg-contaminated floodplain sediments near Oak Ridge, TN.
45 The overall objective of this work is to understand the transport and fate of Hg that enters the surface flow system at the headwaters of Gossan Creek and partitions to the sediments. Using SEM, Leybourne et al. (2000) identified Au– Hg particles, < 1 µm diameter, in the sediment from Gossan Creek and suggested that these formed in the stream system in response to the degradation of Hg– and Au– CN complexes and subsequent reduction of Hg(II) and Au(I) to their elemental forms. One of the specific objectives was to build on previous research by Leybourne et al. (2000) and investigate the mechanisms of Hg mass loss from the stream to the sediments using analytical (S)TEM. Consequently, mineralogical investigations focused on the characterization of the amalgam particles, and identification of additional Hg-bearing secondary mineral phases. The results from this aspect of the study are presented here but additional details about the aqueous and sediment geochemistry and hydrology can be found in Al et al. (2006b).
TEM sample preparation
46 Sediments were sieved to < 45 µm and analyzed for total Hg. A portion of the sample with the highest Hg concentration (1 636 µg/g) was saturated with a low-viscosity epoxy (EPO-TEK 301-2FL) and then pressed into a 1 cm diameter mould to form a pellet. A double-polished thin section was prepared from the pellet and used for SEM– EDS analysis. Mercury-bearing sediment particles were identified with the SEM in the backscattered electron mode. In all cases, these particles were less than 1 to 2 µm in diameter; too small to obtain satisfactory image resolution, or spatial resolution for discrete EDS analyses. For this reason, analytical (S)TEM was used to further investigate the Hg-bearing particles. Samples were prepared for (S)TEM analysis from the standard petrographic thin section using the FIB method described by Patterson et al. (2002).
Summary of results
47 Mercury was identified by SEM– EDS in fine grains (< 2 µm) in association with Au, Ag, Cu and S. Two discrete forms of Hg-bearing mineral grains were observed with (S)TEM– EDS, HgS (Fig. 6) and Hg– Au amalgam with or without Ag. Silver was commonly observed in association with the HgS grains (Fig. 6), suggestive of the presence of a separate Ag-bearing phase, or a Hg– Ag sulphide such as imiterite (Ag2HgS2), but this possibility has not been resolved. Selected-area electron diffraction patterns were collected from the HgS grains whenever possible by tilting the specimen stage to acquire patterns from multiple zones on the same grain. Analysis of the patterns indicates that the HgS is crystalline with a structure consistent with metacinnabar (HgS). This result is consistent with the findings of Barnett et al. (1997) who identified metacinnabar in Hg-contaminated soils using TEM– EDS and electron diffraction, and with the experimental results of Charnock et al. (2003) who used X-ray absorption spectroscopy and XRD to define a sequence of reactions that culminate in the precipitation of stable, crystalline metacinnabar from HS- and Hg(II)-containing solutions. The morphology of the metacinnabar grains presented in Fig. 6A is very delicate, and it is highly unlikely that that these grains could have been derived from mechanical transport from the gossan. We believe that they are secondary precipitates formed in situ within the stream sediments.
48 Analyses of the Hg-bearing amalgam grains suggest that Au is ubiquitous, and the Hg and Ag content of the grains varies widely. Two of these grains are presented in Fig. 7, one with abundant Ag (Figs. 7A, 7B), and the second with virtually no Ag (Figs. 7C-E). The relative proportions of these metals in the amalgam grains are likely to be controlled by their redox speciation and the activity of reduced S species. For example, in SO4 reducing conditions, Hg and Ag are likely to partition to sulphide minerals.
Fig. 6 A) TEM image of metacinnabar grains in stream sediment. The electron diffraction pattern shown in the inset, which is consistent with metacinnabar, is one of five that were collected from the Hg– S-rich portions of the grain aggregate. B) Red– green– blue (RGB) image compiled from separate S (red), Hg (green), and Ag (blue) analytical maps acquired with EDS in STEM mode for the area outlined in A. The orange/yellow areas are HgS (metacinnabar), but the purple indicates a mixture of Ag and S.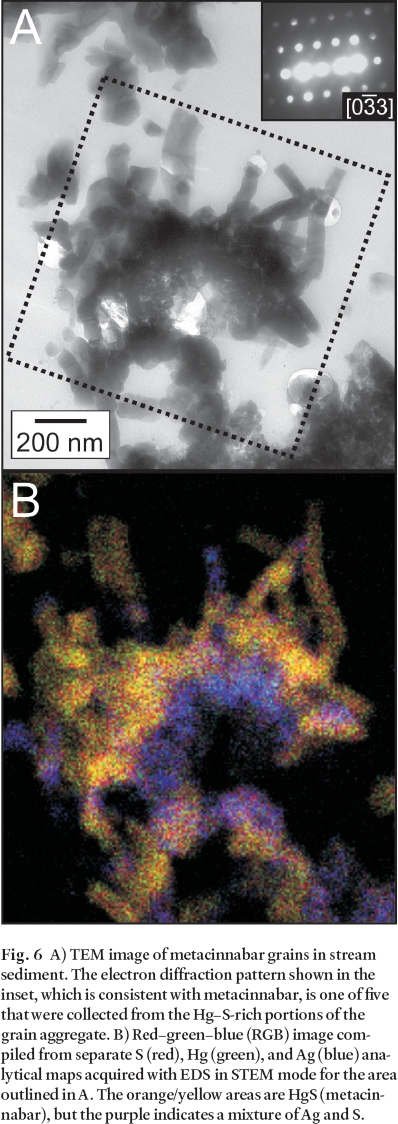
Display large image of Figure 6
49 Abundant sub-micron size Cu-sulphide grains were also noted in the (S)TEM– EDS investigations (Figs. 7A, 7B). Many of them are fine-grained with a delicate morphology (not shown) and, as is the case for the Cu sulphide shown in Fig. 7A, Cu sulphide commonly occurs intergrown with, or encapsulated by the amalgam. Similar to metacinnabar, these grains are thought to have formed in situ within the sediment. The EDS analyses of the grains consistently indicate a high Cu– S ratio which is suggestive of a cuprous sulphide such as chalcocite or djurleite.
50 The results are consistent with Leybourne et al. (2000) in that Hg is attenuated in the stream sediments in the form of Hg– Au amalgam, but it is clear from the (S)TEM analyses that the amalgam is chemically heterogeneous at the nanoscale. For instance, Ag may or may not occur in association with the Hg– Au amalgam. In addition, the results from the (S)TEM analyses indicate that Hg has also partitioned to the sediments in the form of nanoscale metacinnabar. Gold is detected in the form of an amalgam only and Ag occurs in the Hg-, Au-containing amalgam but also in association with Hg and S, perhaps as a discrete Ag or Ag-sulphide phase, or a Hg– Ag-sulphide phase. As a result of the (S)TEM analyses it was also possible to identify the presence of nanoscale secondary Cu minerals in the stream sediments.
Fig. 7 A) BF TEM image showing Au– Ag– Hg amalgam and Cu sulphide. The dark band along the right margin of the grains is tungsten that is deposited as a protective coating prior to the FIB milling procedure. There are relatively small Cu-sulphide particles (light grey) partially surrounding, and enclosed within, the amalgam. B) EDS spectra from the areas indicated in (A). The Ni peaks are background from the TEM sample grid. C) ADF image of a grain of Hg– Au amalgam. The linear features along the right margin arise from a strongly diffracting tungsten coating that penetrated into a crack which was present in the petrographic thin section prior to FIB milling. D) and E) Analytical EDS maps showing the Hg (D) and Au (E) distribution in the amalgam grain presented in (C).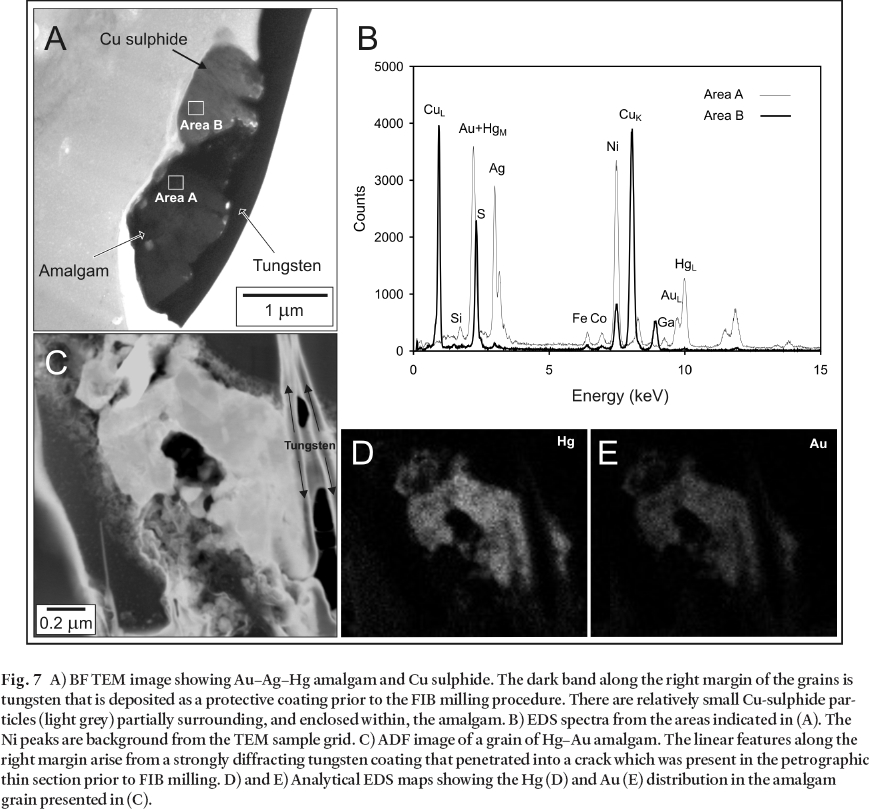
CONCLUSIONS
51 The examples of (S)TEM analyses demonstrate that investigation at the nanoscale can reveal heterogeneity in geologic materials that cannot be adequately resolved at the micron scale. The preparation of geological samples suitable for such analyses no longer poses major problems because of the availability of FIB instruments. Although the examples that were presented here are from low-temperature environments, the preparation and analysis techniques have applications across a broad spectrum of geological studies in all geologic systems.
ACKNOWLEDGEMENTS
The research presented here was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC), Ontario Power Generation (OPG), and the University Consortium for Solvents in Groundwater. The FIB preparation of samples used in the work presented here was done by FIBICS Inc. (Ottawa, ON). The authors wish to thank Dr. D. Hall at the UNB Microscopy and Microanalysis Facility for his assistance with the X-ray microanalysis measurements. The authors would also like to thank the reviewers (Dr. H.E. Jamieson and Dr. D. Paktunc) and the editors of this special issue for providing valuable suggestions and comments.REFERENCES
Al, T.A., Banks, V., Loomer, D., Parker, B.L., & Mayer, K.U. 2006a, Metal mobility during chemical oxidation of TCE by KMnO4. Journal of Contaminant Hydrology, 88, pp. 137– 152.
Al, T.A., Leybourne, M.I., Maprani, A.C., MacQuarrie, K.T., Dalziel, J.A., Fox, D., & Yeats, P.A. 2006b. Effects of acid-sulphate weathering and cyanide-containing gold tailings on the transport and fate of mercury and other metals in Gossan Creek: Murray Brook Mine, New Brunswick, Canada. Applied Geochemistry, 21, pp. 1969– 1985.
Alpers, C.N., & Blowes, D.W. 1994. Environmental Geochemistry of Sulfide Oxidation, ACS Symposium Series 550, American Chemical Society, Washington, DC, 681 p.
Andersen, D.J., Bishop, F.C., & Lindsley, D.H. 1991. Internally consistent solution models for Fe-Mg-Mn-Ti oxides: Fe-Mg-Ti oxides and olivine. American Mineralogist, 76, pp. 417– 444.
Barnett, M.O., Harris, L.A., Turner, R.R., Stevenson, R.J., Henson, T.J., Melton, R.C., & Hoffman, D.P. 1997. Formation of mercuric sulfide in soil. Environmental Science & Technology, 31, pp. 3037– 3043.
Benzerara, K., Yoon, T.H., Menguy, N., Tyliszczak, T., & Brown Jr., G.E. 2005. Nanoscale environments associated with bioweathering of a Mg-Fe-pyroxene. Proceedings of the National Academy of Sciences of the United States of America, 102, pp. 979– 982.
Bezos, A., & Humler, E. 2005. The Fe3+/∑Fe ratios of MORB glasses and their implications for mantle melting. Geochimica et Cosmochimica Acta, 69, pp. 711– 725.
Boyle, D.R. 1995. Geochemistry and genesis of the Murray Brook precious metal gossan deposit, Bathurst mining camp, New Brunswick. Exploration and Mining Geology, 4, pp. 341– 363.
Boyle, D.R., & Smith, C.N. 1994. Mobilization of mercury from a gossan tailings pile, Murray Brook precious metal vat leaching operation. In International Land Reclamation and Mine Drainage Conference and the Third International Conference on the Abatement of Acidic Drainage, New Brunswick, Canada, 4, pp. 254– 263.
Braun, A., Huggins, F.E., Shah, N., Chen, Y., Wirick, S., Mun, S.B., Jacobsen, C., & Huffman, G.P. 2005. Advantages of soft X-ray absorption over TEM-EELS for solid carbon studies— a comparative study on diesel soot with EELS and NEXAFS. Carbon, 43, pp. 117– 124.
Buckley, A.N., & Walker, G.W. 1988. The surface composition of arsenopyrite exposed to oxidizing environments. Applied Surface Science, 35, pp. 227– 240.
Cavé L.C., & Al, T.A. 2005. Paleohydrogeology – development of analytical TEM methods for paleoredox investigations. Ontario Power Generation, Nuclear Waste Management Division Report 06819-REP-01300-10099-R00. 52 p.
Cavé, L., Al, T., Loomer, D., Cogswell, S., & Weaver, L. 2006. A STEM/EELS method for mapping iron valence ratios in oxide minerals. Micron, 37, pp. 301– 309.
Charnock, J.M., Moyes, L.N., Pattrick, R.A.D., Mosselmans, J.F.W., Vaughan, D.J., & Livens, F.R. 2003. The structural evolution of mercury sulfide precipitate: an XAS and XRD study. American Mineralogist, 88, pp. 1197– 1203.
Cressey, G., Henderson, C.M.B., & van der Laan, G. 1993. Use of L-edge X-ray absorption spectroscopy to characterize multiple valence states of 3d transition metals; a new probe for mineralogical and geochemical research. Physics and Chemistry of Minerals, 20, pp. 111– 119.
Crimi, M.L., & Siegrist, R.L. 2004. Impact of reaction conditions on MnO2 genesis during permanganate oxidation. Journal of Environmental Engineering, 130, pp. 562– 572.
Dong, H., Peacor, D.R., & Murphy, S.F. 1998. TEM study of progressive alteration of igneous biotite to kaolinite throughout a weathered soil profile. Geochimica et Cosmochimica Acta, 62, pp. 1881– 1887.
Dobrzhinetskaya, L.F., Green, H.W., Weschler, M., Darus, M., Wang, Y.-C., Massonne, H.-J., & Stöckhert, B. 2003. Focused ion beam technique and transmission electron microscope studies of microdiamonds from the Saxonian Erzgebirge, Germany. Earth and Planetary Science Letters, 210, pp. 399– 410.
Drexel, R.T., Haitzer, M., Ryan, J.N., Aiken, G.R., & Nagy, K.L. 2002. Mercury(II) sorption to two Florida Everglades peats: Evidence for strong and weak binding and competition by dissolved organic matter released from the peat. Environmental Science & Technology, 36, pp. 4058– 4064.
Dyar, M.D., Lowe, E.W., Guidotti, V., & Delaney, J.S. 2002. Fe3+ and Fe2+ partitioning among silicates in metapelites: A synchrotron micro-XANES study. American Mineralogist, 87, pp. 514– 522.
Egerton, R.F. 1996. Electron energy-loss spectroscopy in the electron microscope. 2nd edition. Plenum Press, New York. 485 p.
Fernandez, P.G., Linge, H.G., Wadsley, M.W. 1996a. Oxidation of arsenopyrite (FeAsS) in acid Part I: Reactivity of arsenopyrite. Journal of Applied Electrochemistry, 26, 575– 583.
Fernandez, P.G., Linge, H.G., Willing, M.J. 1996b. Oxidation of arsenopyrite (FeAsS) in acid Part(II): Stoichiometry and reaction scheme. Journal of Applied Electrochemistry, 26, 585– 591.
Frazier, B.E., Wiener, J.G., Rada, R.G., & Engstrom, D.R. 2000. Stratigraphy and historic accumulation of mercury in recent depositional sediments in the Sudbury River, Massachusetts, U.S.A. Canadian Journal of Fisheries and Aquatic Sciences, 57, pp. 1062– 1072.
Garvie, L.A.J., & Buseck, P.R. 1998. Ratios of ferrous to ferric iron from nanometre-sized areas in minerals. Nature, 396, pp. 667– 670.
Gosar, M., Pirc, S., & Bidovec, M. 1997. Mercury in the Idrijca River sediments as a reflection of mining and smelting activities of the Idrija mercury mine. Journal of Geochemical Exploration, 58, pp. 125– 131.
Gray, J.E., Crock, J.G., & Fey, D.L. 2002. Environmental geochemistry of abandoned mercury mines in West-Central Nevada, USA. Applied Geochemistry, 17, pp. 1069– 1079.
Gray, J.E., Hines, M.E., Higueras, P.L., Adatto, I., & Lasorsa, B.K. 2004. Mercury speciation and microbial transformations in mine wastes, stream sediments, and surface waters at the Almadén Mining District, Spain. Environmental Science & Technology, 38, pp. 4285– 4292.
Heaney, P.J., Vicenzi, E.P., Giannuzzi, L.A., & Livi, K.J.T. 2001. Focused ion beam milling: A method of site-specific sample extraction for microanalysis of Earth and planetary materials. American Mineralogist, 86, pp. 1094– 1099.
Hitchcock, A.P., Morin, C., Zhang, X., Araki, T., Dynes, J., Stöver, H., Brash, J., Lawrence, J.R., Leppard, G.G. 2005. Soft X-ray spectromicroscopy of biological and synthetic polymer systems. Journal of Electron Spectroscopy and Related Phenomena, 144-147, pp. 259– 269.
Hochella Jr., M.F. 2002a. There's plenty of room at the bottom: Nanoscience in geochemistry. Geochimica et Cosmochimica Acta, 66, pp. 735– 743.
Hochella Jr., M.F. 2002b. Nanoscience and technology: the next revolution in the Earth sciences. Earth and Planetary Science Letters, 203, pp. 593– 605.
Holdaway, M.J., Mukhopadhyay, B., Dyar, M.D., Guidotti, C.V., & Dutrow, B.L. 1997. Garnet-biotite geothermometry revised: New Margules parameters and a natural specimen data set from Maine. American Mineralogist, 82, pp. 582– 595.
Huang, K.-C., Hoag, G.E., Chheda, P., Woody, B.A., & Dobbs, G.M. 2002. Chemical oxidation of trichloroethylene with potassium permanganate in a porous medium. Advances in Environmental Research, 7, pp. 217– 229.
Jackson, J.M., Sturhahn, W., Shen, G., Zhao, J., Hu, M.Y., Errandonea, D., Bass, J.D., & Fei, Y. 2005. A synchrotron Mössbauer spectroscopy study of (Mg,Fe)SiO3 perovskite up to 120 GPa. American Mineralogist, 90, pp. 199– 205.
Jambor, J.L., & Blowes, D.W. 1994. Short Course Handbook on Environmental Geochemistry of Sulfide Mine-Wastes, Volume 22. Mineralogical Association of Canada, Nepean, ON, 438 p.
Jambor, J.L., Blowes, D.W., & Ritchie, A.I.M. 2003. Environmental Aspects of Mine Wastes, Short Course Series, Volume 31, Mineralogical Association of Canada, Nepean, ON, 430 p.
Jeong, G.Y., & Kim, H.B. 2003. Mineralogy, chemistry, and formation of oxidized biotite in the weathering profile of granitic rocks. American Mineralogist, 88, pp. 352– 364.
Jones, R.A., Koval, S.F., & Nesbitt, H.W. 2003. Surface alteration of arsenopyrite (FeAsS) by Thiobacillus oxidans Geochimica et Cosmochimica Acta, 67, pp. 955– 965.
Kanungo, S.B., Tripathy, S.S., Mishra, S.K., Sahoo, B., & Rajeev. 2004. Adsorption of Co2+, Ni2+, Cu2+, and Zn2+ onto amorphous hydrous manganese dioxide from simple (1– 1) electrolyte solutions. Journal of Colloid and Interface Science, 269, pp. 11– 21.
Kim, C.S., Rytuba, J.J., & Brown Jr., G.E. 2004a. EXAFS study of mercury(II) sorption to Fe- and Al-(hydr)oxides I. Effects of pH. Journal of Colloid and Interface Science, 271, pp. 1– 15.
Kim, C.S., Rytuba, J.J., & Brown Jr., G.E. 2004b. EXAFS study of mercury(II) sorption to Fe- and Al-(hydr)oxides II. Effects of chloride and sulfate. Journal of Colloid and Interface Science, 270, pp. 9– 20.
Lechler, P.J., Miller, J.R., Hsu, L.-C., & Desilets, M.O. 1997. Mercury mobility at the Carson River Superfund Site, west-central Nevada, USA: interpretation of mercury speciation data in mill tailings, soils, and sediments. Journal of Geochemical Exploration, 58, pp. 259– 267.
Lee, M.R., Bland, P.A., & Graham, G. 2003. Preparation of TEM samples by focused ion beam (FIB) techniques: applications to the study of clays and phyllosilicates in meteorites. Mineralogical Magazine, 67, pp. 581– 592.
Lemon, L.A., & Pheeney, P. 1996. Hydrogeological study-1996, Murray Brook Mine site, Murray Brook Mine, NB., Jacques Whitford and Associates Limited, Fredericton, NB. 54 p.
Leybourne, M.I., Goodfellow, W.D., Boyle, D.R., & Hall, G.E.M. 2000. Form and distribution of gold mobilized into surface waters and sediments from a gossan tailings pile, Murray Brook massive sulphide deposit, New Brunswick, Canada. Applied Geochemistry, 15, pp. 629– 646.
Li, X.D., & Schwartz, F.W. 2004. DNAPL remediation with in situ chemical oxidation using potassium permanganate. Part I. Mineralogy of Mn oxide and its dissolution in organic acids. Journal of Contaminant Hydrology, 68, pp. 39– 53.
Loomer, D.B., Al, T.A., Weaver, L., & Cogswell, S. In press. Manganese valence imaging in Mn minerals at the nanoscale using STEM-EELS. American Mineralogist, 92, pp. 72– 79.
Maprani, A.C., Al, T.A., MacQuarrie, K.T., Dalziel, J.A., Shaw, S.A., & Yeats, P.A. 2005. Determination of mercury evasion in a contaminated headwater stream. Environmental Science & Technology, 39, pp. 1679– 1687.
McCanta, M.C., Dyar, M.D., Rutherford, M.J., & Delaney, J.S. 2004. Iron partitioning between basaltic melts and clinopyroxene as a function of oxygen fugacity. American Mineralogist, 89, pp. 1685– 1693.
McMurry, J., & Ejeckam, R.B. 2002. Paleohydrogeological study of fracture mineralogy in the Whiteshell Research Area. Ontario Power Generation, Nuclear Waste Management Division Report 06819-REP-01200-10082-R00. 81 p.
Mikhlin, Yu., & Tomashevich, Ye. 2005. Pristine and reacted surfaces of pyrrhotite and arsenopyrite as studied by X-ray absorption near-edge structure spectroscopy. Physics and Chemistry of Minerals, 32, pp. 19– 27.
Nelson, M.D., Parker, B.L., Al, T.A., Cherry, J.A., & Loomer, D. 2001. Geochemical reactions resulting from in-situ oxidation of PCE-DNAPL by KMnO4 4 in a sandy aquifer. Environmental Science & Technology, 35, pp. 1266– 1275.
Nesbitt, H.W., Muir, I.J., & Pratt, A.R. 1995. Oxidation of arsenopyrite by air and air-saturated, distilled water, and implications for mechanism of oxidation. Geochimica et Cosmochimica Acta, 59, pp. 1773– 1786.
Nesbitt, H.W., & Muir, I.J. 1998. Oxidation states and speciation of secondary products on pyrite and arsenopyrite reacted with mine waste waters and air. Mineralogy and Petrology, 62, pp. 123– 144.
Patterson, R.J., Mayer, D., Weaver, L., & Phaneuf, M.W. 2002. "H-bar lift-out" and "Plan-view lift-out": Robust, rethinnable FIB-TEM preparation for ex-situ cross-sectional and plan-view FIB specimen preparation. Microscopy and Microanalysis, 8 (Supplement 2), pp. 566– 567.
Petrunic, B.M., & Al, T.A. 2005. Mineral/water interactions in tailings from a tungsten mine, Mount Pleasant, New Brunswick. Geochimica et Cosmochimica Acta, 69, pp. 2469– 2483.
Petrunic, B.M., Al, T.A., & Weaver, L. 2006. A transmission electron microscopy analysis of secondary minerals formed in tungsten-mine tailings with an emphasis on arsenopyrite oxidation. Applied Geochemistry, 21, pp. 1259– 1273.
Pretorius, P.J., & Linder, P.W. 2001. The adsorption characteristics of ä-manganese dioxide: a collection of diffuse double layer constants for the adsorption of H+, Cu2+, Ni2+, Zn2+, Cd2+ and Pb2+. Applied Geochemistry, 16, pp. 1067– 1082.
Raeburn, S.P., Ilton, E.S., & Veblen, D.R. 1997. Quantitative determination of the oxidation state of iron in biotite using X-ray photoelectron spectroscopy: II. In situ analyses. Geochimica et Cosmochimica Acta, 61, pp. 4531– 4537.
Richardson, S., & Vaughan, D.J. 1989. Arsenopyrite: a spectroscopic investigation of altered surfaces. Mineralogical Magazine, 53, pp. 223– 229.
Rightor, E.G., Hitchcock, A.P., Ade, H., Leapman, R.D., Urquhart, S.G., Smith, A.P., Mitchell, G., Fischer, D., Shin, H.J., & Warwick, T. 1997. Spectromicroscopy of poly(ethylene terephthalate): Comparison of spectra and radiation damage rates in X-ray absorption and electron energy loss. Journal of Physical Chemistry B, 101, pp. 1950– 1960.
Schaufuss, A.G., Nesbitt, H.W., Scaini, M.J., Hoechst, H., Bancroft, M.G., & Szargan, R. 2000. Reactivity of surface sites on fractured arsenopyrite (FeAsS) toward oxygen. American Mineralogist, 85, pp. 1754– 1766.
Schumacher, J.C. 1991. Empirical ferric iron corrections: necessity, assumptions, and effects on selected geothermo-barometers. Mineralogical Magazine, 55, pp. 3– 18.
Seydoux-Guillaume, A.-M., Goncalves, P., Wirth, R., & Deutsch, A. 2003. Transmission electron microscope study of polyphase and discordant monazites: Site-specific specimen preparation using the focused ion beam technique. Geology, 31, pp. 973– 976.
Shaw, S.A., Al, T.A., & MacQuarrie, K.T.B. In press. Mercury mobility in unsaturated gold mine tailings, Murray Brook mine, New Brunswick, Canada. Applied Geochemistry, 21, pp. 1986– 1998.
Tonkin, J.W., Balistrieri, L.S., & Murray, J.W. 2004. Modeling sorption of divalent metal cations on hydrous manganese oxide using the diffuse double layer model. Applied Geochemistry, 19, pp. 29– 53.
van Aken, P.A., Liebscher, B., & Styrsa, V.J. 1998. Quantitative determination of iron oxidation states in minerals using Fe L2,3 -edge electron energy-loss near-edge structure spectroscopy. Physics and Chemistry of Minerals, 25, pp. 323– 327.
van Aken, P.A., & Liebscher, B. 2002. Quantification of ferrous/ferric ratios in minerals: new evaluation schemes of FeL2,3 electron energy-loss near-edge spectra. Physics and Chemistry of Minerals, 29, pp. 188– 200.
Williams, D.B., & Carter, C.B. 1996. Transmission Electron Microscopy, A Textbook for Materials Science, Plenum Press, New York, 729 p.
Williams, H.M., McCammon, C.A., Peslier, A.H., Halliday, A.N., Teutsch, N., Levasseur, S., & Burg, J.-P. 2004. Iron isotope fractionation and the oxygen fugacity of the mantle. Science, 304, pp. 1656– 1659.
Wirth, R. 2004. Focused Ion Beam (FIB): A novel technology for advanced application of micro- and nanoanalysis in geosciences and applied mineralogy. European Journal of Mineralogy, 16, pp. 863– 876.
Wu, C.-M., Zhang, J., & Ren, L.-D. 2004. Empirical garnet-muscovite-plagioclase-quartz geobarometry in medium- to high-grade metapelites. Lithos, 78, pp. 319– 332.
Editorial responsibility: Michael B. Parsons