Special Series - Enviromental Geoscience
Impact of historical gold mining activities on marine sediments in Wine Harbour, Nova Scotia, Canada
ABSTRACT
Past investigations at historical gold (Au) districts in Nova Scotia, Canada have identified elevated concentrations of arsenic (As) and mercury (Hg) in nearby sediments and waters. These metal(loid)s are derived from erosion of mineralized bedrock, and the disposal of mine tailings into the environment during early operations. The Wine Harbour gold district is located along the eastern shore of Nova Scotia, and produced 1329 kg of Au from 75 581 tonnes of crushed rock from 1862 to 1939. The gold occurs in arsenopyrite-bearing quartz-carbonate veins and was extracted using stamp milling and Hg amalgamation. Historical maps document tailings deposits near former stamp mill sites; however, the extent to which these mine wastes influence environmental quality in the adjacent marine environment is uncertain. In this study, we measured metal(loid) concentrations in tailings, marine sediments, and surface waters to assess the lateral and vertical extent of mining-related impacts on Wine Harbour. Chemical analyses of terrestrial and intertidal tailings reveal high concentrations of both As (86–196 000 mg/kg) and Hg (444–320 000 µg/kg). Analyses of marine sediments show a wide range in both As (4–568 mg/kg) and Hg (<5–7430 µg/kg) concentrations. In general, the highest metal(loid) concentrations in sediments were recorded down-gradient of stamp mill sites. Elevated concentrations were also detected in sediments underlying an active mussel aquaculture operation at the western end of the harbour. Results from this study have been used to help assess potential ecosystem and human health risks associated with historical gold mine wastes in the Wine Harbour area.
RÉSUMÉ
Les investigations antérieures réalisées dans les anciens champs d’or (Au) de la Nouvelle-Écosse, au Canada, ont mis en évidence des concentrations élevées d’arsenic (As) et de mercure (Hg) dans les eaux et les sédiments avoisinants. Ces éléments chimiques non métalliques proviennent de l’érosion du substratum minéralisé et des résidus miniers rejetés dans l’environnement au début des activités d’exploitation. De 1862 à 1939, le champ d’or de Wine Harbour, situé le long de la côte est de la Nouvelle-Écosse, a produit 1 329 kg d’Au à partir de 75 581 tonnes de pierres concassées. Présent dans des filons de quartz-carbonates riches en arsénopyrite, l’or a été extrait au moyen de techniques de bocardage et d’amalgamation au Hg. Les cartes historiques font état de dépôts de résidus à proximité des sites de bocardage. Cependant, la mesure dans laquelle ces déchets miniers influent sur la qualité environnementale du milieu marin adjacent est incertaine. Dans le cadre de la présente étude, nous avons mesuré les concentrations d’éléments chimiques non métalliques dans les résidus miniers, les sédiments marins et les eaux de surface afin d’évaluer l’étendue latérale et verticale des incidences de l’exploitation minière sur Wine Harbour. Les analyses chimiques des résidus miniers terrestres et intertidaux révèlent de fortes concentrations d’As (de 86 mg/kg à 196 000 mg/kg) et de Hg (de 444 µg/kg à 320 000 µg/kg). Les analyses des sédiments marins indiquent que les concentrations d’As (de 4 mg/kg à 568 mg/kg) et de Hg (moins de 5 µg/kg à 7 430 µg/kg) varient grandement. En général, les plus fortes concentrations d’éléments chimiques non métalliques ont été enregistrées en aval des sites de bocardage. De fortes concentrations ont aussi été détectées dans les sédiments situés au-dessous d’une exploitation mytilicole active à l’extrémité ouest du havre. Les résultats de cette étude ont été utilisés pour aider à évaluer les risques que les déchets des anciennes mines d’or dans la région de Wine Harbour pourraient présenter pour l’écosystème et la santé humaine.
[Traduit par la redaction]
INTRODUCTION
1 Historical gold production on mainland Nova Scotia occurred within 64 districts from 1861 to the mid-1940s (Bates 1987). These early operations relied primarily on Hg amalgamation to extract Au from ore, and it is estimated that 10–25% of the initial Hg added during milling was routinely lost to the environment (Henderson 1935; Nriagu and Wong 1997). Moreover, As occurs naturally in minerals (e.g., arsenopyrite, FeAsS) associated with the auriferous host rock, and was concentrated in mine waste (tailings) through mineral processing activities. In the absence of environmental regulations during historical operations, tailings were generally discharged directly onto the surrounding land and into receiving environments including wetlands, streams, lakes, and the ocean. Investigations dating back to the 1970s have identified elevated concentrations of As and Hg in sediments, waters, vegetation, and biota downstream of these gold districts (e.g., Environmental Protection Service 1978; Brooks et al. 1982; Dale and Freedman 1982; Mudroch and Clair 1986; Wong et al. 1999, 2002; Moriarty et al. 2009, 2013; Saunders et al. 2010; Parsons et al. 2012). However, the fate and effects of historical gold mine tailings disposed directly into the marine environment from coastal stamp mills are not well understood.
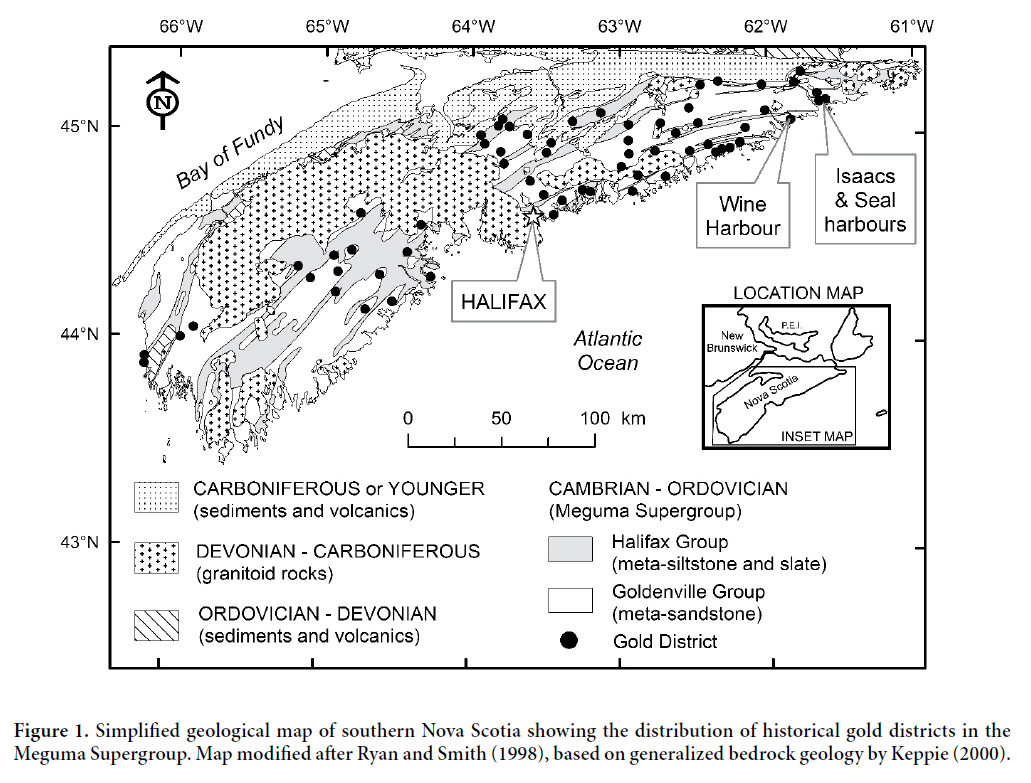
2 Many of the gold districts in Nova Scotia are located along the coast, or are drained by streams and rivers that flow directly to the ocean. In the Isaacs Harbour and Wine Harbour districts (Fig. 1), there are several former stamp mill sites along the shoreline with no associated on-land tailings deposits, suggesting that most of the wastes from these mills were discharged into the ocean. In 2004, research by Natural Resources Canada revealed widespread contamination of the intertidal zone in Seal Harbour (Fig. 1) by tailings derived from the abandoned Lower Seal Harbour gold mine, located 2.3 km inland (Parsons et al. 2012). Follow-up studies of soft-shelled clams collected from an intertidal tailings flat in Seal Harbour documented some of the highest concentrations of As in clam tissues ever reported in the scientific literature (up to 2300 ppm dry weight; K. Doe, Environment Canada, personal communication, 2005), leading to the closure of this area for bivalve shellfish harvesting in May 2005 (Environment Canada 2007). Subsequent investigations have shown that high concentrations of As and Hg are associated with loosely packed aggregates of particulate material (flocs) near the sediment-water interface in Seal Harbour, which are readily available for ingestion by filter-feeding organisms (Milligan and Law 2013). Detailed studies of As speciation in clams, blue mussels, periwinkles, and seaweed collected from Seal Harbour reveal that much of the As in these edible species is in a highly toxic inorganic form, which may pose a risk to consumers (Koch et al. 2007; Whaley-Martin et al. 2012, 2013; Doe et al. in press).
3 The widespread occurrence and innocuous appearance of historical gold tailings deposits in Nova Scotia create the potential for human exposure, and recent land-use changes in some districts have increased these risks (Parsons et al. 2012). The only documented case of human health impacts took place in 1976, when a resident living near a past-producing gold district (Waverley) was diagnosed with chronic arsenic intoxication after drinking well water containing high levels of As derived from tailings and waste rock deposits (Grantham and Jones 1977). Since 2003, Natural Resources Canada and its partners have carried out extensive studies of the Nova Scotia gold mine districts to characterize the distribution, chemical form, and potential risks associated with metal(loid)s derived from these historical mine wastes (Parsons et al. 2012). These studies have documented large volumes of unconfined tailings in many gold districts, with some deposits extending more than 2 km from their source. Early results from these investigations led to the formation of the Provincial-Federal Historic Gold Mines Advisory Committee in 2005. This committee has evaluated ecological and human health risks at selected gold mine sites throughout Nova Scotia, coordinated research on tailings management strategies, and advised Nova Scotian residents on how to reduce their exposure to gold mine tailings (Nova Scotia Environment 2015).
4 In this study, we use field observations and geochemical analyses to characterize the lateral and vertical extent of gold mine tailings in the marine and terrestrial environments at the past-producing Wine Harbour gold district in Nova Scotia. Quantitative data for As and Hg burdens in mine tailings, marine sediments, and water are interpreted in the context of data reported for 14 other historical gold districts on mainland Nova Scotia. The overall objective of this investigation is to provide geoscience data that can be used to assess and mitigate future risks from gold mine wastes in the Wine Harbour area.
SITE DESCRIPTION
5 The Wine Harbour gold district is located in Guysborough County, on the eastern shore of mainland Nova Scotia (Fig. 1). Gold-bearing veins are hosted in rocks of the Cambro-Ordovician Meguma Supergroup, which is subdivided into the Goldenville and Halifax groups (White 2010). The basal Upper Cambrian Goldenville Group is composed of metamorphosed sandstone, siltstone, and shale, which are conformably overlain by Ordovician slate, shale, siltstone, and minor sandstones of the Halifax Group (Smith et al. 2005). Accessory sulphide phases are abundant in both groups, and include pyrite, pyrrhotite, and arsenopyrite (Kontak and Jackson 1999). Auriferous deposits at Wine Harbour are hosted primarily in the Goldenville Group, and mineralization is closely associated with the southwest-trending Wine Harbour anticline. Smith et al. (1985) have identified saddle-reef gold deposits in the hinges of anticlinal folds at Wine Harbour, as well as six different vein types including stratiform, stratabound, side, step, discordant and vertical. The vein mineralogy consists mainly of quartz ± chlorite ± carbonate ± K-feldspar ± muscovite ± sulphides. Further details on the geology of the Wine Harbour gold district can be found in Little (2006).
 Display large image of Figure 1
Display large image of Figure 1
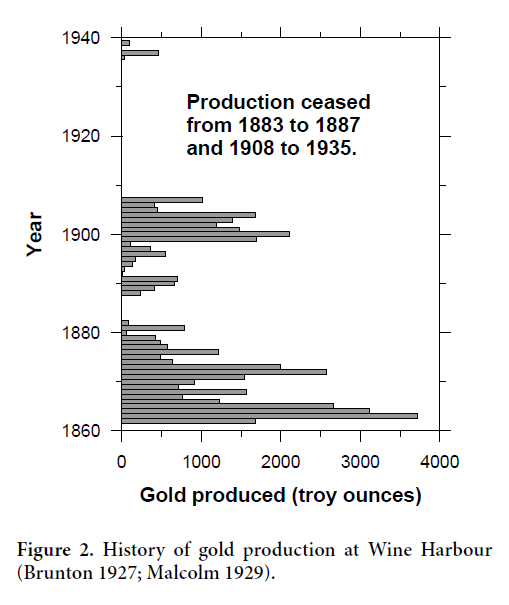
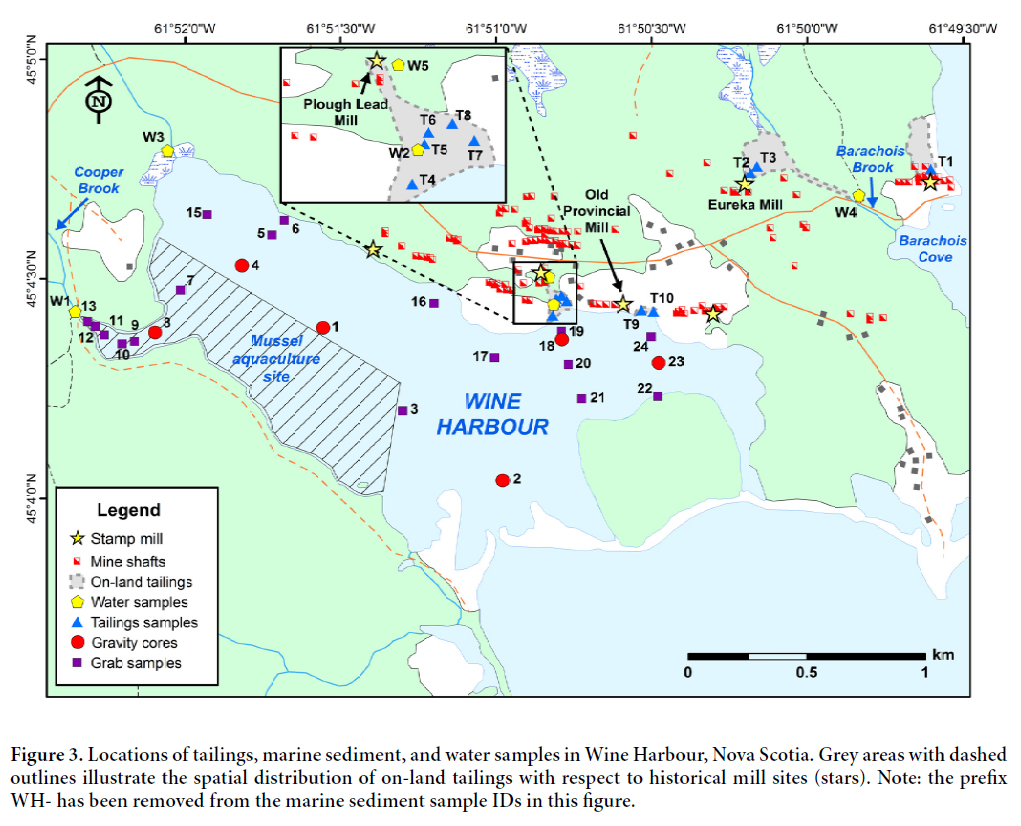
6 Mining of quartz-carbonate veins at Wine Harbour took place between 1862 and 1907 and again from 1936 to 1939, yielding 1329 kg (42 727 troy oz.) of Au from 75 581 tonnes of crushed rock (Fig. 2) (Malcolm 1929; Nova Scotia Department of Mines 1961). Ore milling using Hg-amalgamation was conducted on-site at six main stamp mills distributed throughout the district (Faribault 1905). Since production ceased in 1939, there has been no environmental remediation within the district, and tailings deposits persist in the area. Field investigations conducted by Seabright Resources in the early 1980s concluded that most tailings deposits are located in close proximity to old stamp mill structures (Glover et al. 1983). Their observations suggest that the majority of tailings were located on land, with a lesser amount stretching out short distances into the harbour in areas where mills were adjacent to the shoreline. Tailings deposits were also documented in freshwater streams, particularly Barachois Brook, where the Eureka Mining Company had constructed a 10-stamp mill (Fig. 3) (Glover et al. 1983). Currently, the on-land tailings are heavily overgrown by spruce, fir, alders, and other mixed shrubs.
 Display large image of Figure 2
Display large image of Figure 2
 Display large image of Figure 3
Display large image of Figure 3
METHODS
Field Sampling
Tailings sampling
7 Fourteen on-land tailings samples were collected from ten sites (Fig. 3), and included surficial samples, and samples of discrete horizons at depth. Sites were selected based on their proximity to old stamp mill structures using a detailed Geological Survey of Canada map of the Wine Harbour gold district as a guide (Faribault 1905). In general, dug pits were 30 to 40 cm wide by 20 to 50 cm deep, and tailings were collected using a stainless steel trowel and plastic sampling containers. Sampling tools were thoroughly cleaned between sites to prevent cross-contamination between samples. The number of samples collected at each site was governed by the location and vertical stratigraphy of the tailings deposit. At sites with well-defined down-hole variations in colour, samples were typically collected from both the oxidized and unoxidized layers. At water-logged sites, one sample from the unoxidized layer was considered sufficient. Full descriptions of each sample site can be found in Appendix A of Little (2006).
Marine sediment sampling
8 Marine sediments were collected from 23 separate locations, at sites distributed throughout the harbour (Fig. 3). The location of these sites was partially dictated by the shallow nature of the harbour, which limited access to some areas. Sample sites located down-gradient of near-shore tailings deposits were of particular interest, as these were considered to have an increased likelihood of hosting submarine tailings accumulations. A higher density of samples were collected within an operational mussel aquaculture site at the western end of Wine Harbour to check for elevated As and Hg concentrations in sediments within the site boundaries. Marine sediment samples included 17 surface sediments and six sediment cores (Fig. 3), collected using a Wildco® Ekman grab sampler and a 50 lb Benthos® gravity corer, respectively. In order to minimize sediment disturbance a core catcher was not used during collection of the sediment cores. Cores were 2.5 inches (6.35 cm) in diameter and ranged from 42 to 56 cm in length.
Water sampling
9 Five surface water samples were collected from four streams in the Wine Harbour area, three of which drain directly into Wine Harbour and a fourth (W4) at Barachois Brook (Fig. 3), which is downstream of the Eureka mill, and drains into neighbouring Indian Harbour. Sample sites were selected to investigate the release of As and Hg from historical mill sites, with the exception of site W3, which is located at the outlet of a small, unnamed brook near the western end of the harbour in an area with no documented historical mining activity. Sample site W1 was located at Cooper Brook, which drains into a small inlet at the western end of the harbour and is downstream of a former cyanide plant that was used for reprocessing tailings (Malcolm 1929). Sample site W2 was located in a small stream that passes directly through an existing tailings deposit down-slope from the historical Plough Lead Mining Company’s mill. Sample W5 was collected approximately 120 m upstream from site W2, where this stream originates from a collapsed adit beneath the Plough Lead mill. Surface water samples were collected during a single sampling event (July 19, 2005), which was conducted under low-flow conditions.
10 In situ measurements of pH, temperature and specific conductance were taken at each site prior to water sampling using a miniLab IQ125 pH meter and a Corning Checkmate II conductivity meter, respectively. Five 60-mL water samples were collected from each of the sites, using acid-washed, high-density polyethylene containers (Nalgene® 2114-0002). Each batch consisted of two filtered (Sterivex™ capsule filters with a <0.45 µm Durapore membrane) and two unfiltered water samples, designated for cation and Hg analyses. A fifth sample was collected for measurements of anions, dissolved organic carbon (DOC), and alkalinity. Travel blanks, acid blanks, and sample blanks were also collected to check for possible contamination during sample processing. Samples collected for Hg analysis were preserved with 0.5 mL of ultrapure bromine chloride (BrCl), and samples designated for cation analysis were preserved using 0.5 mL of 8 N ultrapure nitric acid (HNO3). All samples were stored at 4°C prior to shipment to the Analytical Method Development labs at the Geological Survey of Canada, in Ottawa, Ontario, where the analyses were completed within 90 days.
Laboratory analyses
Tailings composition and mineralogy
11 Tailings were subsampled and freeze-dried at the Geological Survey of Canada (Atlantic) (GSC-A) in Dartmouth, Nova Scotia prior to bulk chemical analyses at ACME Analytical Laboratories in North Vancouver, British Columbia. A 1.0 g portion of each subsample (<2 mm size fraction) was digested in 6 mL of a 1:1:1 solution of HCl:HNO3:H2O at a temperature of 95°C for one hour (Package IF-MS). All samples were analyzed for 53 elements using inductively coupled plasma - mass spectrometry (ICP-MS; Thermo X7 Series II). Samples with concentrations of As and/or Hg greater than the upper limit of the 1F-MS package were re-analyzed using ACME’s 7AR Multi-Element Assay by Inductively Coupled Plasma - Emission Spectrometry (ICP-ES) protocol following aqua regia digestion. Accuracy and precision of analytical procedures were monitored through the submission of duplicate samples, and certified reference materials STSDs 2-4 (Lynch 1990) and NIST 1646 (National Bureau of Standards 1982). In general, analytical accuracy and precision were within ± 5 to 10% of the expected values for most elements except Hg, which showed some evidence of carry over in certified reference materials that were run immediately following samples with very high Hg concentrations. Careful analysis of results from duplicate samples shows little evidence for this carry-over effect and it is not expected to affect the conclusions of our study. Total carbon content of tailings and sediment samples was measured in 0.5 g sub-samples at GSC-A using a LECO WR-112 carbon analyser. Organic carbon concentrations were analysed following removal of the inorganic carbon using 1 M HCl. Inorganic carbon concentrations were calculated by difference. Precision and accuracy were approximately ± 0.05 wt.% based on replicate analyses of calibration standards.
12 Bulk mineralogical analyses of 14 tailings samples were conducted via X-ray powder-diffraction (XRD) using a Siemens D–500 X-ray diffractometer with a Co lamp (18 mA, 20 keV). Each sample was finely ground using an agate mortar and pestle, then run from 2° to 70° 2θ, in steps of 0.02°, with a 1 s count time at each step. Six of these samples were further characterized using electron probe microanalyses (EPMA) to quantitatively define mineral chemistry and to help identify potential sources of As and Hg. Grain mounts were analyzed using wavelength dispersive spectrometry (WDS), on a JEOL 8200 microanalyzer at Dalhousie University in Halifax, Nova Scotia. Calibration standards used for EPMA included cinnabar (Hg), sanidine (K), apatite (Ca) and arsenopyrite (As, S, Fe). Arsenopyrite was also used as an analytical control for these analyses.
Marine sediment geochemistry, geochronology, and grain size
13 Marine sediment cores were vertically extruded using a calibrated polycarbonate extruder, and subsampled at 1 cm intervals down to a depth of 30 cm. Beyond this depth, intervals were increased to a thickness of 2 cm. Core subsamples were homogenized and subdivided for radionuclide, grain size, and elemental analyses. Grab samples were subsampled in the field by collecting a single sample from the surface (0–1 cm) using a stainless steel spatula. In instances where multiple layers were evident in the grab sample, a second subsample was collected at depth (~10 cm). All core and grab subsamples were collected away from the edges of the sampler. Samples were freeze-dried and shipped to ACME Analytical Labs for elemental analyses. Bulk geochemical and carbon analyses followed the same protocol as previously detailed for the tailings samples.
14 Natural (210Pb) and artificial (137Cs) radionuclide tracers in two marine sediment cores were used to reconstruct historical trends of metal input to Wine Harbour. These cores were selected because they showed the least evidence of mixing at the sediment-water interface. Lead-210 activities in freeze-dried subsamples were determined by alpha counting of the granddaughter nuclide 210Po which was electrodeposited from the dissolved sediment sample onto nickel discs from weak acid solution. Both 137Cs and the background (supported) 210Pb activity, derived from in-situ decay of 226Ra in the sediment, were measured by direct counting of dried sediment samples using a hyper-pure Ge gamma ray spectrometer. Excess 210Pb (the quantity transported through the water column) was then determined by the difference between the total and 226Ra supported 210Pb activities (Smith and Walton 1980).
15 Sedimentation rates and geochronologies were determined from the 210Pb and 137Cs sediment depth distributions using previously outlined methods (Smith et al. 2009). The sedimentation rate was calculated from the slope of a least squares exponential fit of excess 210Pb as a function of sediment depth and mass-depth (g/cm2). The geochronology was then estimated from the 210Pb-determined sedimentation rate. This methodology assumes that both the flux of excess 210Pb through the water column and the sedimentation rate have remained constant over the past 100 years. Cesium-137 was plotted on the geochronological scale determined from the 210Pb sedimentation rate. Generally, the 137Cs threshold horizon and maximum should be in agreement with 210Pb dates of approximately 1952 and 1964 corresponding to the shape of the 137Cs fallout record (Smith et al. 2009). The absence of this agreement may indicate that the core has undergone mixing by processes such as bioturbation or be otherwise undatable (Smith 2001). The 137Cs distributions in the present cores were consistent with fallout profiles predicted from the 210Pb sedimentation rates (WH-04, 0.371 cm/y; WH-23, 0.389 cm/y) and geochronologies indicating that sedimentation patterns have been uniform in these depositional regimes of Wine Harbour during the latter half of the 20th century.
16 The disaggregated inorganic grain size (DIGS) distributions of bottom sediment samples were determined using a Coulter Multisizer IIe. Organic matter was first removed from bottom sediments with 35% hydrogen peroxide (H2O2). Prior to analysis on the Multisizer, inorganic sediments were resuspended in a 1% NaCl solution then disaggregated with a sapphire-tipped ultrasonic probe. Three tubes, with apertures of 30 200 and 400 µm, were used to obtain the DIGS distribution over a size range from 0.8 to 240 µm. Particle sizes were binned in a 1/5 phi interval (φ = -log2D; D= diameter in mm). The DIGS obtained from the Multisizer are expressed as log of equivalent volume fraction versus log of the diameter. Volume fraction is assumed to equal mass fraction, which implies constant particle density across all sizes. Uncertainty in DIGS analysis is <10%, which is the error in the Coulter Multisizer IIe. For a complete description of the methods used for particle size analysis, see Kranck and Milligan (1979) and Milligan and Kranck (1991).
17 The inverse floc model of Curran et al. (2004) applies a non-linear least squares fit to the DIGS curves. Floc fraction, Kf, is the mass fraction of material that was deposited to the seabed in flocs and can be described by the equation:
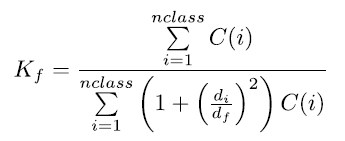
where C(i) is the concentration of size class i, nclass represents the number of size classes present in the DIGS distribution, and di is the diameter of particles in class i. Floc fraction in the sediments represents an integration of f, the mass fraction of flocs in suspension, over multiple tidal cycles. Error introduced in fitting the model to the DIGS distributions is assumed to be less than that introduced from error in Coulter Counter measurements (i.e., <10%) and therefore does not affect the parameter values. For a complete background theory of the inverse model see Kranck et al. (1996), Curran et al. (2004), and Law et al. (2013).
Water chemistry
18 Surface waters were analyzed for cations, anions, Hg and DOC concentrations at the Analytical Method Development labs at GSC-Ottawa. Major element concentrations were measured using inductively coupled plasma-atomic emission spectrometry (ICP-AES; PerkinElmer model 3000 DV), while minor and trace element concentrations were quantified by ICP-MS (Thermo X7 Series II). Mercury concentrations were analyzed using a Tekran 2600 Hg analyzer (detection limit, 0.5 ng/L). Anion concentrations were determined via a Dionex DX-600 ion chromatograph. High-temperature combustion was used to determine the dissolved organic carbon content of all water samples, following the removal of inorganic carbon using H3PO4. Alkalinity measurements were completed via automated Gran titrations. The complete analytical dataset can be found in Little (2006).
RESULTS AND DISCUSSION
Locations of tailings deposits
19 Field identification of on-land tailings deposits was based on their colour, vertical stratigraphy, and fine-grained texture. In Wine Harbour, contrasting depositional environments (i.e., terrestrial versus intertidal) have produced variably oxidized deposits, which exhibit distinct down-hole appearances. In drier environments, tailings typically display a vertical colour gradient ranging from rusty brown at the surface to dark grey at depth as conditions become anoxic (Fig. 4a). In waterlogged areas along the shoreline of Wine Harbour, the extent of oxidation displayed in tailings deposits is less variable. Here, tailings maintain a light- to dark-grey colour throughout as a result of reduced oxygen penetration at the surface (Fig. 4b).
 Display large image of Figure 4
Display large image of Figure 4
20 Field observations and geochemical results (discussed below) confirmed previous assumptions that four main tailings deposits occupy the terrestrial and marine environments at Wine Harbour. Based on historical maps and ruins observed in the field, the distribution of these deposits is generally consistent with the locations of former mill structures (Fig. 3), and they are largely confined to the northeastern part of the district. Two deposits are located along the shoreline, directly down-gradient of the Plough Lead and Old Provincial mill sites. Northeast of these deposits, a third accumulation is located down-gradient of the Old Eureka mill, and a fourth is situated just east of this in a swampy area close to Barachois Cove.
Tailings chemistry
21 Arsenic and Hg concentrations measured in subsamples from 10 on-land tailings sites at Wine Harbour (Table 1) are consistent with ranges reported for 14 other historical gold districts in Nova Scotia (Parsons et al. 2012). These tailings contain As concentrations ranging between 200 and 196 000 mg/kg (median 4880 mg/kg), and comparison with the Canadian environmental quality guideline for As in soils (12 mg/kg; Canadian Council of Ministers of the Environment (CCME 2001)) demonstrate that As burdens are exceedingly high. Mercury concentrations range from 4930 to 320 000 µg/kg (median 11 300 µg/kg), and 90% of these samples have concentrations in excess of the CCME Hg soil quality guideline (6600 µg/kg) (CCME 1999). Field observations, conventional XRD, and EPMA results confirm that the anomalous concentration of As (196 000 mg/kg) measured in sample T5B is caused by the abundance of arsenopyrite (Little 2006). Furthermore, the markedly high concentration of Hg (320 000 µg/kg) measured in T5B suggest that it may represent a sulphide-rich mill concentrate mixed with tailings. By comparison, concentration ranges for As and Hg measured in tailings and stream sediments from other gold districts in Nova Scotia are 10 mg/kg to 310 000 mg/kg (median 2550 mg/kg) and <5 µg/kg to 350 000 µg/kg (median 1640 µg/kg), respectively (Parsons et al. 2012).
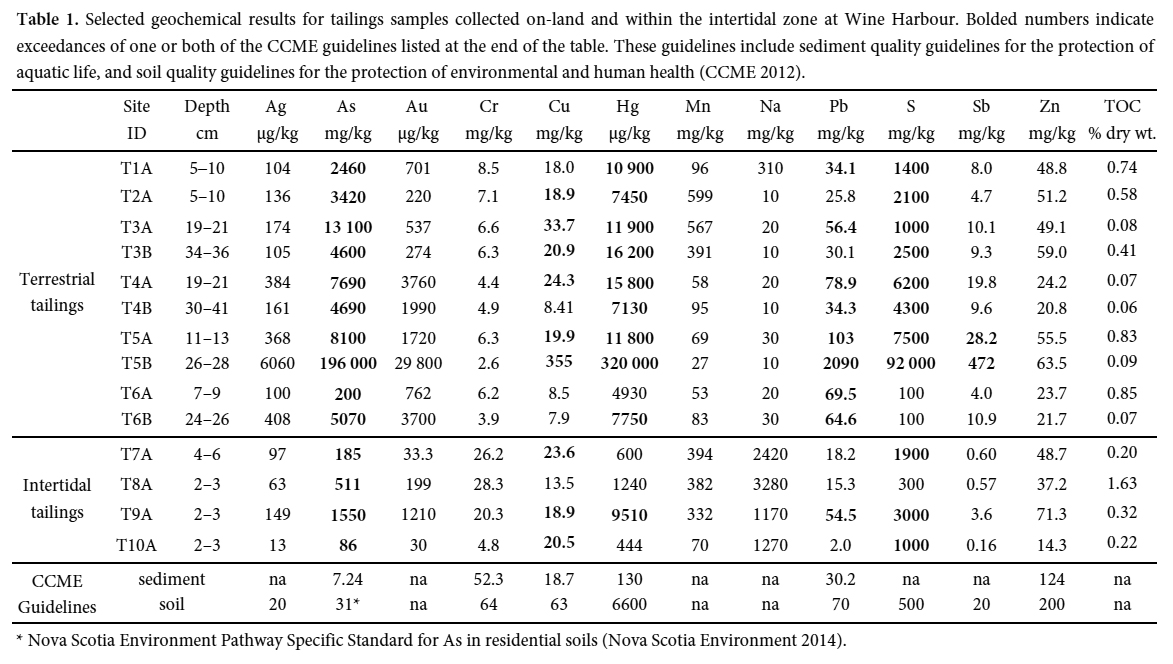 Display large image of Table 1* Nova Scotia Environment Pathway Specific Standard for As in residential soils (Nova Scotia Environment 2014).
Display large image of Table 1* Nova Scotia Environment Pathway Specific Standard for As in residential soils (Nova Scotia Environment 2014).
22 Intertidal zone tailings (n = 4) sampled down-gradient of two near-shore mill sites contain concentrations of As and Hg that are considerably lower than tailings located above the high water line. Here, As ranges between 86 and 1550 mg/kg (median 348 mg/kg), and all sites have As concentrations in excess of the CCME 12 mg/kg soil quality guideline. In contrast, Hg ranges from 444 to 9510 µg/kg (median 922 µg/kg), and only one site has concentrations in excess of the 6600 µg/kg guideline. The comparatively low concentrations of As and Hg in the intertidal zone probably reflect mixing of the tailings with natural marine sediments, as well as settling of dense sulphide minerals (e.g., arsenopyrite) close to the original stamp mill sites.
23 The concentrations of As and Hg in the tailings and intertidal sediments at Wine Harbour are significantly higher than the naturally occurring levels of As and Hg in soils overlying mineralized bedrock within the Nova Scotia gold districts. Parsons and Little (2015) reported As and Hg concentrations in near surface (0–5 cm, <2 mm size fraction) soils collected from 85 sites at the Montague and Goldenville gold districts to be 2–273 mg/kg (median 23 mg/kg) and 39–490 µg/kg (median 137 µg/kg), respectively. The elevated As and Hg burdens documented in the tailings and intertidal zone samples at Wine Harbour suggest that they could present a risk to human and ecosystem health (CCME 1999, 2001).
Tailings mineralogy
24 The major mineralogy of 14 terrestrial tailings samples is consistent with studies conducted at other Nova Scotia gold districts, and dominant minerals reflect auriferous vein and host-rock compositions (Walker et al. 2009; Corriveau et al. 2011; DeSisto et al. 2011; Percival et al. 2014). Primary minerals identified through bulk XRD analyses include quartz, plagioclase feldspar, chlorite, muscovite, and graphite, with secondary Fe-oxide phases (goethite) probable in some samples. Moreover, qualitative energy-dispersive spectrometry (EDS) analyses using EPMA identified various accessory phases, including arsenopyrite, pyrite, pyrrhotite, ilmenite, zircon, and gold. EPMA photomicrographs and backscattered electron images in Little (2006) illustrate the contrast in mineralogical compositions between terrestrial tailings deposits and those in the intertidal marine environment. Most notably, Little (2006) observed an absence of sulphide grains at intertidal locations. This pattern implies that the tailings were heavily diluted upon entry into the harbour, and/or sulphide oxidation or settling is prevalent in shallow marine sediments. Elevated concentrations of sodium (Na, 1170–3280 mg/kg) and boron (B, 1–9 mg/kg) measured in the intertidal samples reflect compositional differences between near-shore and terrestrial samples (Harder 1970; Chester 2000).
25 Quantitative EPMA determined arsenopyrite to be the primary As-bearing phase present in Wine Harbour tailings. Under reduced conditions, the reactivity of sulphides such as arsenopyrite and pyrite is limited, which is illustrated by the absence of reaction rims observed in photomicrographs. In contrast, sulphides in well-drained terrestrial environments exhibited variable states of oxidation, producing secondary phases such as Fe-oxides and Ca-Fe arsenates near surface. The element map in Fig. 5a illustrates this by highlighting the distribution of As, Hg, iron (Fe) and sulphur (S) in an arsenopyrite grain in the Plough Lead mill concentrate sample. This sample was panned from within the foundation of the Plough Lead mill, and consists of a mixture of grey, Fe-rich spheres, visible gold, and Fe-stained, sand-size material. Here, the element map exhibits a distinct Fe-arsenate reaction rim formed at the edge of the grain as a result of sulphide oxidation. An area of elemental Hg was also identified in the same sample (Fig. 5b) and provides direct evidence of Hg loss during amalgamation.
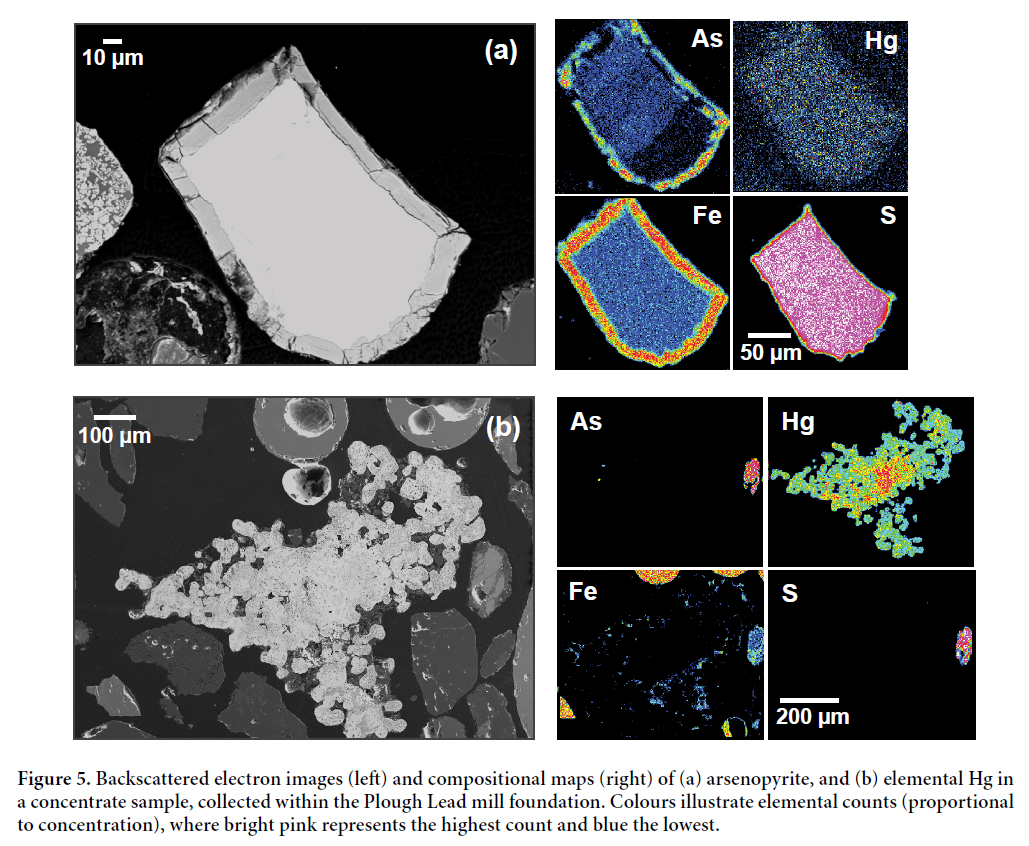 Display large image of Figure 5
Display large image of Figure 5
Marine sediment chemical composition
26 Bulk chemical analyses of sediments captured in grab samples and gravity cores indicate that marine sediments in Wine Harbour were heavily influenced by the deposition of historical mine tailings. The ranges of As (4–568 mg/kg) and Hg (<5–7430 µg/kg) concentrations (Table 2) measured in Wine Harbour marine sediments are considerably larger than those reported in bottom sediments from 10 inlets and harbours along the Atlantic coastline of Nova Scotia (As 2–42 mg/kg, n = 157; Hg 10–390 µg/kg, n = 180; Loring et al. 1996). District-scale maps highlight three distinct areas of elevated As and Hg in marine sediments situated down-gradient of the Plough Lead and Old Provincial mill sites at the eastern end of the harbour, and to the west in a small inlet at the mouth of Cooper Brook (Figs. 6 a, b). These maps illustrate the average As and Hg concentrations in grab samples, and the top 15 cm of gravity cores. Reported concentrations in sediment cores are restricted to the top 15 cm on the basis of the ecological significance of these near-surface sediments, as the zone of active bioturbation in marine sediments extends to a mean global depth of 9.7 ± 4.5 cm (Boudreau 1998).
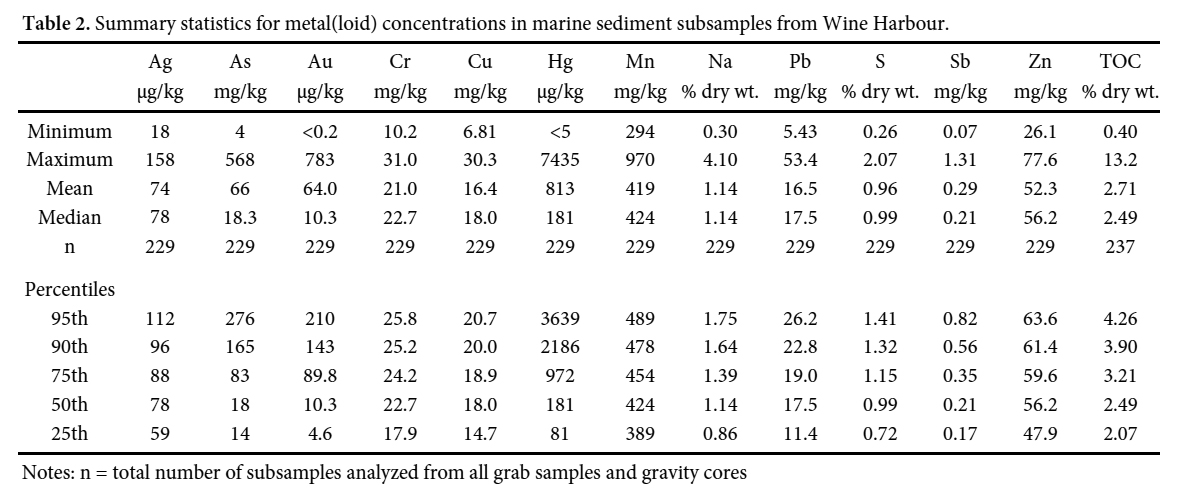 Display large image of Table 2Notes: n = total number of subsamples analyzed from all grab samples and gravity cores
Display large image of Table 2Notes: n = total number of subsamples analyzed from all grab samples and gravity cores
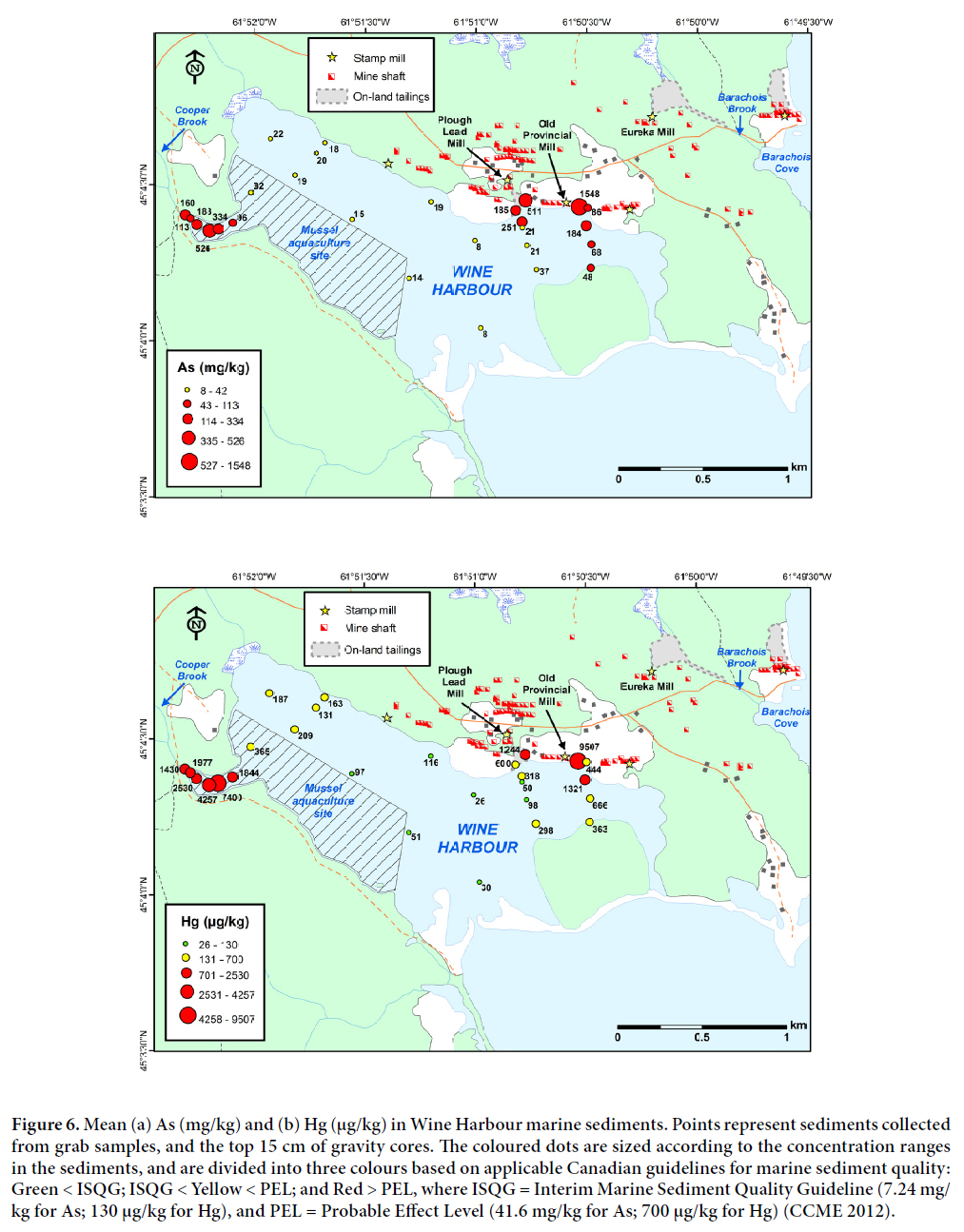 Display large image of Figure 6
Display large image of Figure 6
27 Dispersal patterns suggest that adjacent terrestrial tailings deposits are the main source of As and Hg to the marine sediments (Figs. 6 a, b). An exception to this includes the small inlet at the western end of the harbour, which also hosts a mussel aquaculture site. Early accounts by Malcolm (1929) indicate that in 1899, a cyanide plant was used to reprocess historical mine tailings on Cooper Brook, which drains directly into this inlet. However, this brook is not included on historical maps of the area (Faribault 1905), and no cyanide plant structural remains were identified in the field due to the dense cover of vegetation. Thus, definitive identification of the sources of As and Hg to the marine environment in this area will rely on future investigations. Overall, the footprint of elevated As and Hg concentrations in Wine Harbour is likely the combined product of the direct disposal of mine wastes into the harbour between 1862 and 1939, and the subsequent reworking of these contaminated sediments by marine processes.
28 Vertical sediment profiles for each of the gravity cores collected at Wine Harbour showed variations in As, Hg, and other elemental concentrations with depth. Paired with radionuclide tracer data (210Pb, 137Cs), chemical profiles for two of these cores (WH-04 and WH-23; Fig. 3) illustrate the chronology of metal contamination in Wine Harbour marine sediments (Fig. 7). Lead-210 and 137Cs data for these two cores showed the least evidence of mixing near the sediment-water interface and should therefore provide the most robust geochronologies for the sediments in Wine Harbour. Overall, As and Hg profiles for all sediment cores show diminishing concentrations with increasing distance from historical mill sites. Metal(loid) concentrations measured in deep (>25 cm) bottom sediments located near the harbour’s entrance are relatively constant with depth and likely reflect background conditions that pre-date historical gold production. In core WH-02 (Fig. 3), the estimated ranges in naturally occurring As and Hg values are 4–6 mg/kg (median 5 mg/kg) and Hg 5–17 µg/kg (median 11 µg/kg), respectively. These background levels are below the Canadian Interim Marine Sediment Quality Guidelines for both As (7.24 mg/kg) and Hg (130 µg/kg) (CCME 2012). By comparison, As and Hg burdens in cores collected throughout most other areas of Wine Harbour remain above these background values down to a depth of at least 40 cm, with the highest As and Hg concentrations (up to 568 mg/kg and 6860 µg/kg, respectively) observed in sediments from cores WH-08 and WH-23.
 Display large image of Figure 7
Display large image of Figure 7
29 Chronologically, the peaks in As and Hg concentrations in marine sediment cores correspond roughly with documented periods of mining and milling activity at Wine Harbour (Fig. 2). This pattern is especially evident in core WH-23 (Fig. 7a), where As, Au, and Hg profiles exhibit marked increases above background concentrations between a depth of 50 and 37 cm below the sediment-water interface, before decreasing to lower concentrations in shallower sediments. Lead-210 and 137Cs data for this core indicate that the rapid increase in As, Au, and Hg concentrations at about 45 cm coincides with an increase in the amount of ore crushed in the late 1880s, and the subsequent drop in concentrations above 37 cm reflects the sudden halt in production in 1907 (Fig. 2). None of the cores collected during this study were of sufficient length to document the environmental impact of the earliest periods of gold production at Wine Harbour between 1862 and 1880. The down-core profiles of As, Au, and Hg also show that the concentrations of these elements remained elevated in the sediments long after mining activity had ceased in the Wine Harbour gold district, and that the near-surface baseline values of As, Au, and Hg are higher than pre-mining background concentrations.
30 Some cores collected from Wine Harbour also show pronounced peaks in As and Hg concentrations near the sediment-water interface (top 0–1 cm), followed by abrupt decreases in metal burdens directly below. For example, core WH-04 (Fig. 7b) was collected from the western end of the harbour (Fig. 3) and contains near-surface concentrations of 47 mg/kg As and 556 µg/kg Hg, as well as distinct peaks in the concentrations of other elements including cadmium, copper, gold, molybdenum, lead, silver, and sulphur (Little 2006). As discussed by Milligan and Law (2013), many contaminants in aquatic environments are associated with loosely packed aggregates of material called flocs, which consist of colloids, organic material, and small inorganic particles that accumulate at the sediment-water interface. This reactive material has a high specific surface area and is ideally suited to accumulate and transport contaminants in the marine environment. The presence of this floc layer is likely responsible for the spike in metalloid concentrations at the surface of core WH-04, and it may represent an important pathway for the uptake of contaminants by filter-feeding organisms (Koch et al. 2007; Whaley-Martin et al. 2012, 2013; Milligan and Law 2013).
31 The controls on flocculation include particle encounter rate, which depends nonlinearly on concentration, particle contact efficiency and stickiness, and particle break-up due to turbulence (Hill 1996; Milligan and Law 2005; Hill et al. 2013). Large particle concentrations and increases in particle “stickiness” associated with increased organic matter, in combination with low-to-moderate turbulent shear, will increase flocculation rates, which in turn increases the settling velocity and associated vertical flux of fine grained material to the seabed. Naturally occurring flocs have settling velocities on the order of 1 mm s-1 (Hill et al. 2001; Fox et al. 2004; Curran et al. 2007) and can actively scavenge metals and organic contaminants and rapidly deposit them to the seabed (Milligan and Loring 1997). Downcore observations of the floc fraction in core WH-23 from 37 cm to 50 cm depth, in conjunction with grain size and trace element data, show increases in both the floc fraction and As and Hg concentrations at a time when grain size decreases (Fig. 8). The median diameter of the sediments (d50), calculated from the DIGS, dropped from approximately 16 μm in the upper 30 cm of the core to roughly 13 μm from 37 cm to 49 cm down-core. This increase in fine-grained sediment loading corresponds to the period of increased gold production between the late 1800s and 1907 (Fig. 2), when large volumes of tailings were deposited in Wine Harbour.
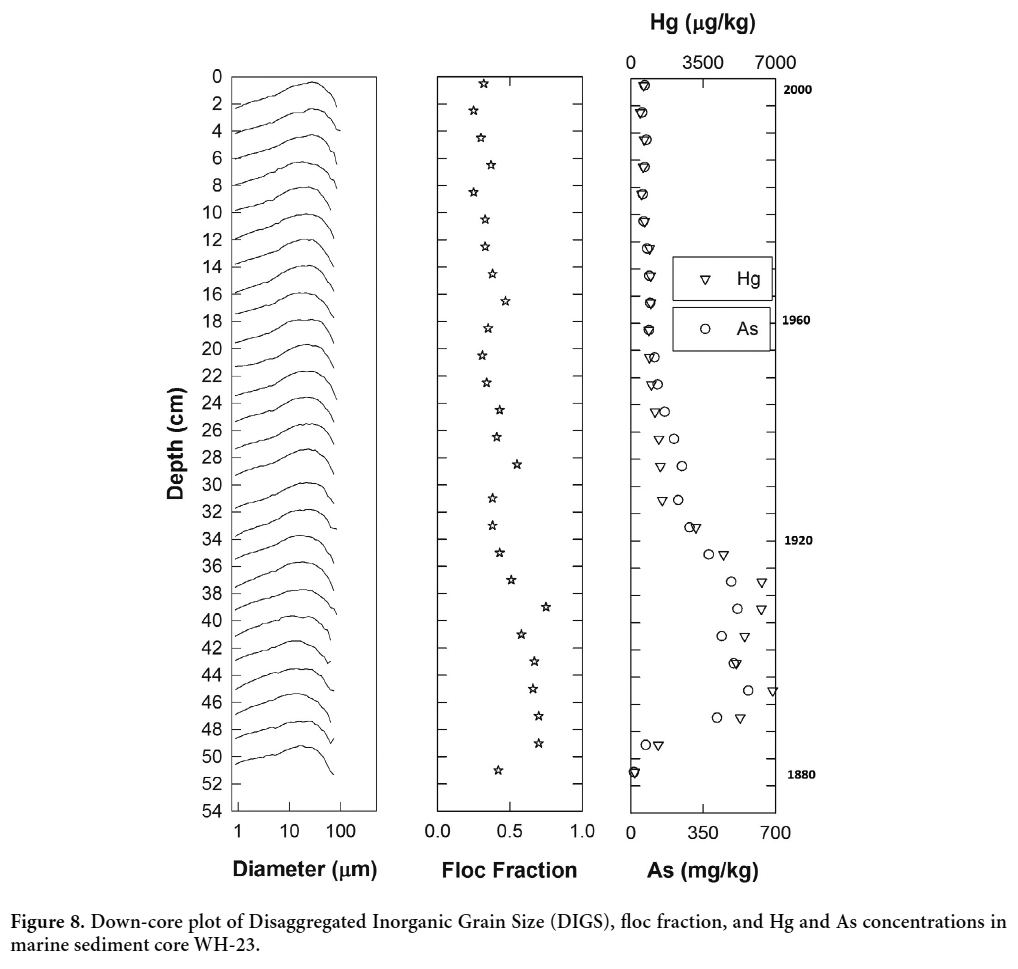 Display large image of Figure 8
Display large image of Figure 8
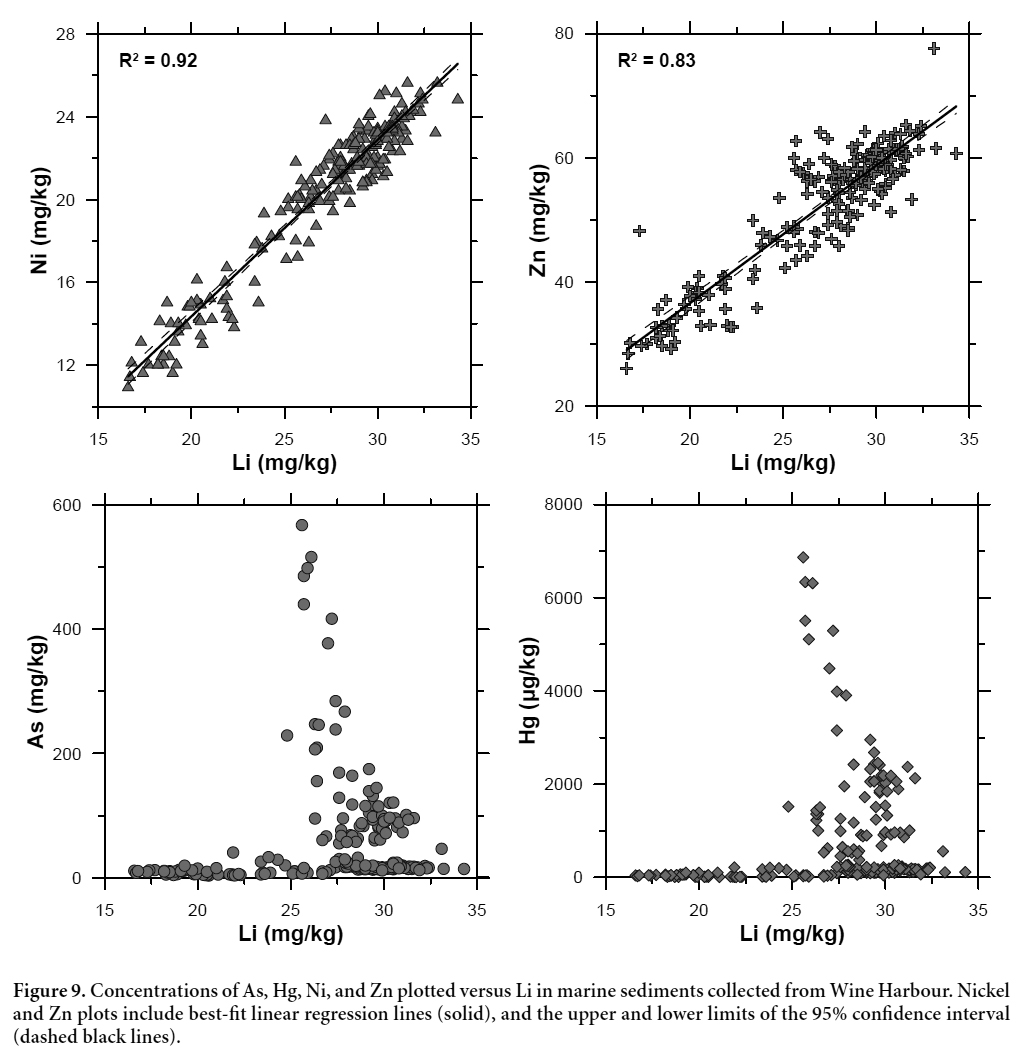
32 When interpreting the relative contributions of metal(loid)s from both natural and anthropogenic sources in marine sediments, it is important to consider the natural variability in sediment mineralogy and grain size distribution (Parsons and Cranston 2006). Normalization of metal concentrations against an element of geogenic origin can correct for such variations, and help to distinguish between natural and anthropogenic enrichments (Summers et al. 1996; Covelli and Fontolan 1997). In this study, we used lithium (Li) as a geochemical proxy for variations in sediment grain size, as it is a common component of clay minerals and micas in glaciomarine sediments, is not affected by post-depositional diagenetic remobilization, and is strongly correlated with changes in grain size in the marine sediments of eastern Canada (Loring 1991). Figure 9 shows the concentrations of As, Hg, nickel (Ni), and zinc (Zn) plotted against Li for all marine sediment subsamples (n = 210). The strong correlation between Li and Ni (r2 = 0.92) and Li and Zn (r2 = 0.83) demonstrates that changes in sediment grain size are the predominant control on the concentrations of these elements in Wine Harbour (cf. Loring 1991). In contrast, plots for As and Hg versus Li show no correlation between the two elements and Li. This lack of correlation confirms that As and Hg concentrations in the Wine Harbour sediments are largely controlled by an external source; in this case, the historical disposal and subsequent redistribution of gold mine tailings.
33 Figure 10 shows the concentrations of As and Hg in marine sediments collected from 14 coastal inlets along the southern and eastern shores of mainland Nova Scotia, including Wine Harbour and inlets near two neighbouring historical gold districts (Isaacs and Seal harbours; Fig. 1). The As concentrations in sediments near all three gold districts are much higher than levels reported for other coastal inlets in Nova Scotia, including industrialized embayments such as Lunenburg Harbour and the LaHave estuary (As data are not available for Halifax Harbour). The Hg burdens in sediments from Wine Harbour are particularly high, reflecting the continuous use of Hg amalgamation for the duration of gold production in this district. The comparatively lower Hg burdens in tailings-contaminated sediments from Seal Harbour reflect the extensive use of cyanidation during the last period of gold mining operations in the Lower Seal Harbour district in the late 1930s (Parsons et al. 2012). The high concentrations of Hg measured in Halifax Harbour sediments are representative of areas contaminated by domestic sewage and effluents from hospitals, dental offices, and industrial sources (Williams 2010).
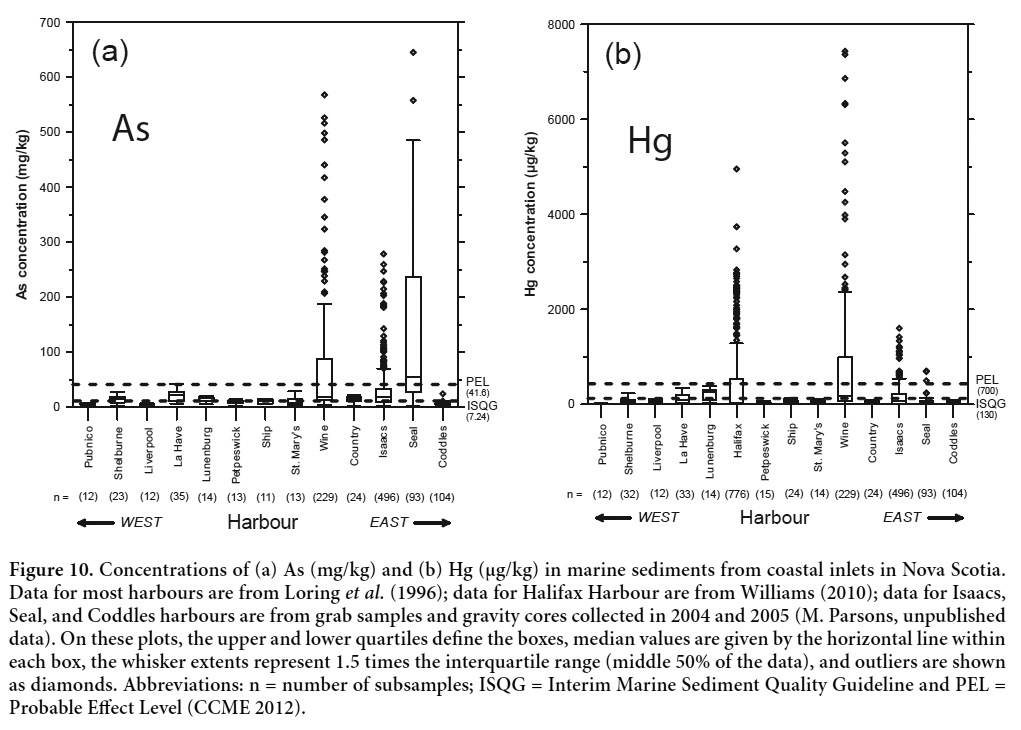 Display large image of Figure 10
Display large image of Figure 10Water Chemistry
34 Samples of stream water and mine drainage collected throughout the Wine Harbour gold district show relatively little contamination from historical gold mining operations under low-flow conditions. Table 3 shows selected water quality data from the five sampling sites labeled in Fig. 3. These results represent water compositions during a single sampling event on July 19, 2005, thus do not account for seasonal fluctuations inherent in some chemical parameters. Water pH remained relatively constant across sites, with a range of 5.4 to 5.8. The temperature of the three stream waters (W1, W3, W4) was warm (18–20 °C), whereas the mine drainage flowing from the collapsed adit beneath the Plough Lead mill (W2, W5) was distinctly cooler (6.9–8.6 °C) and had higher specific conductance.
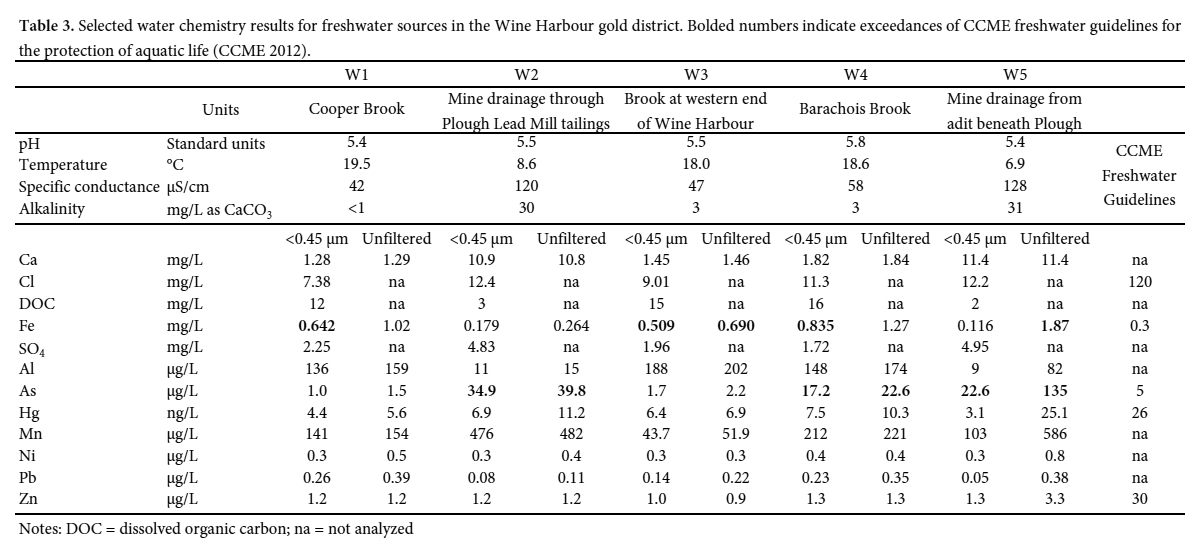 Display large image of Table 3
Display large image of Table 335 All stream waters sampled in the Wine Harbour district exhibit relatively low concentrations of As and Hg. Dissolved concentrations (<0.45 µm) range from 1.0 to 17 µg/L and 4.4 to 7.5 ng/L for As and Hg, respectively. Total (unfiltered) concentrations measure between 1.5 and 23 µg/L for As, and 5.6 and 10 ng/L for Hg. Overall, most of the As and Hg in these freshwater streams is present in dissolved form, or as colloids <0.45 µm in diameter. As expected, the mine drainage originating beneath the Plough Lead Mill (W5) contains higher concentrations of dissolved (23 µg/L) and total As (140 µg/L), reflecting the abundance of arsenopyrite in the underground mine workings. Drainage from this portal also contains the highest total Hg concentration (25 ng/L) of all samples collected, which may be related to the extensive use of Hg in the Plough Lead Mill (Fig. 5b). As this mine drainage passes through the tailings at Site W2, the proportion of As and Hg in the dissolved phase increases and the total concentrations of each element decrease before entering Wine Harbour.
36 In general, dissolved As concentrations in Wine Harbour surface waters are considerably lower than those measured in streams and rivers impacted by mine tailings at 14 other historical gold districts in Nova Scotia. Parsons et al. (2012) reported a range of 0.2 to 6580 µg/L dissolved As (median 117 µg/L), with the highest concentrations associated with standing water on tailings surfaces and in streams running directly through tailings deposits. Background concentrations in mineralized, but unmined, catchments within these gold districts were generally <25 µg/L, which is consistent with the three stream water samples measured in this study. In contrast, dissolved Hg concentrations in surface waters from Wine Harbour are consistent with those measured in surface waters sampled in mined catchments at other historical gold districts (Parsons et al. 2012). Dissolved Hg in waters from the 14 other districts ranged from 1.2 to 61 ng/L. Even in close proximity to tailings deposits, dissolved Hg generally remained below 25 ng/L, suggesting that most Hg in the tailings is present in relatively insoluble forms (e.g., elemental mercury, amalgams, metacinnabar; Parsons et al. 2012).
37 In summary, the relatively low concentrations of As and Hg in stream waters and mine drainage suggest that these freshwater sources are not a major source of contamination to Wine Harbour under relatively low-flow conditions. Additional work would be required to assess the seasonal variations and effects of storms on As and Hg concentrations in the streams that drain into Wine Harbour. Concurrent sampling of marine water from 14 sites in Wine Harbour in July 2005 showed that the dissolved concentrations of As (0.75–1.4 µg/L) and Hg (0.17–4.1 ng/L) are also relatively low in the harbour water (P. Yeats, Fisheries and Oceans Canada, personal communication, 2006). These results are consistent with the work of Doe et al. (in press), who demonstrate that bioaccumulation of As and Hg by organisms living in direct contact with contaminated sediments is the primary risk to ecosystem and human health in the Wine Harbour gold district.
CONCLUSIONS
38 Mercury amalgamation techniques, unconfined tailings disposal, and post-depositional erosion of contaminated sediments are responsible for the release of high concentrations of naturally-occurring As and anthropogenically-derived Hg to the terrestrial and marine environments in the historical Wine Harbour gold district. The highest concentrations of As and Hg are found in on-land tailings deposits in close proximity to historical mill sites near the eastern end of the harbour. Upon entering the harbour, mine tailings were diluted and mixed with natural marine sediments, resulting in lower metal(loid) concentrations in the marine environment. However, in many areas As and Hg burdens in sediments still exceed CCME guidelines for the protection of marine aquatic life. In vertical marine sediment profiles, the chronology of As and Hg contamination closely matches the history of gold production in this district between 1862 and 1939. Comparison of As and Hg concentrations in sediments from pre- and post-mining periods shows that recent sediments are still enriched in metal(loid)s relative to background values as a result of ongoing remobilization of contaminated sediments in Wine Harbour.
39 Unlike sediment, freshwater sources at Wine Harbour sampled in July 2005 show minimal contamination from historical mining activities. This suggests that As and Hg inputs from local streams are not of significant environmental concern under low-flow conditions. The highest As and Hg concentrations were measured in mine drainage from a collapsed adit beneath the Plough Lead Mill, which crosses an overgrown tailings deposit before entering the harbour. Natural streams throughout the district show low As and Hg concentrations that fall within the range of background concentrations measured in unmined catchments at other historical gold districts in Nova Scotia.
40 This investigation was conducted as part of a multidisciplinary study of the environmental impacts of historical gold mining operations throughout Nova Scotia. Our results provide the first direct evidence of the disposal of gold mine tailings into Wine Harbour, and have delineated the spatial extent of this contamination. When combined with information from concurrent biological investigations, these results can be used to help minimize health risks and inform land-use decisions in the Wine Harbour area.
This work was originally conducted as part of the lead author’s Honours B.Sc. thesis in the Department of Earth Sciences at Dalhousie University in Halifax, Nova Scotia. Financial support was provided by the Metals in the Environment (MITE) and Geoscience for Oceans Management (GOM) programs, Earth Sciences Sector, Natural Resources Canada. The authors are grateful to Anne-Marie Ryan (Dalhousie University) for her helpful suggestions throughout this study, and to Brian Fisher and Paul Smith (Nova Scotia Department of Natural Resources) for their contributions to GIS map production and research guidance, respectively. Furthermore, we wish to thank Gordon Brown and Patricia Stoffyn (Dalhousie University), as well as William LeBlanc and Judy Vaive (GSC) for their technical assistance in the laboratory. The manuscript was improved through constructive reviews by Jennifer Galloway, Jeanne Percival, Greg Piorkowski, and Tony Walker, who are acknowledged with thanks. This is contribution number 20150150 of the Earth Sciences Sector, Natural Resources Canada.